| Issue |
Cah. Myol.
Number 25, juillet 2022
|
|
|---|---|---|
| Page(s) | 6 - 9 | |
| Section | Mise au point / Focus | |
| DOI | https://doi.org/10.1051/myolog/202225003 | |
| Published online | 11 août 2022 | |
Les myosites à éosinophiles idiopathiques
Idiopathic eosinophilic myositis
1
Département de Médecine Interne et Immunologie Clinique Centre Hospitalo-Universitaire Edouard Herriot Hospices Civils de Lyon, France
2
Université Paris Est-Créteil, Inserm IMRB U955, Créteil, France Centre de référence des maladies neuromusculaires Centre Hospitalo-Universitaire Henri-Mondor Créteil, France
* Contact Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Résumé
Les myosites à éosinophiles appartiennent au groupe des myopathies inflammatoires idiopathiques et sont définies par un infiltrat inflammatoire musculaire composé de polynucléaires éosinophiles. Il n’existe pas à ce jour de consensus concernant le diagnostic et le traitement de ces patients. Grâce à une revue exhaustive de la littérature, les principales caractéristiques cliniques et histologiques, ainsi que le traitement et l’évolution des patients, ont été résumés dans cette synthèse. Cette revue a permis de distinguer trois sous-groupes de myosites à éosinophiles : la forme focale, la forme diffuse et les périmyosites à éosinophiles. Un algorithme de traitement et de prise en charge est proposé, et les principaux diagnostics différentiels sont discutés.
© L. Gallay et al., publié par EDP Sciences, 2022
 Cet article est distribué sous licence « Creative Commons » : http://creativecommons.org/licenses/by/4.0/deed.fr/, permettant une ré-utilisation du contenu sans restriction à condition de mentionner clairement la source.
Cet article est distribué sous licence « Creative Commons » : http://creativecommons.org/licenses/by/4.0/deed.fr/, permettant une ré-utilisation du contenu sans restriction à condition de mentionner clairement la source.
Introduction
Les premières descriptions de myosite à éosinophiles remontent aux années 1950 avec les premières mentions d’infiltrats d’éosinophiles au sein de muscles masticateurs des chiens. Layzer et al. [1] ont rapporté des cas similaires chez l’homme en 1977, mais cette pathologie reste rare et peu rapportée. Les myosites à éosinophiles appartiennent au groupe des myopathies acquises inflammatoires idiopathiques et sont définies par un infiltrat inflammatoire musculaire composé de polynucléaires éosinophiles, parfois associé à une éosinophilie sanguine. D’un point de vue histologique, le seuil de 0,3 éosinophiles par mm2 dans l’infiltrat inflammatoire musculaire a été proposé par certains auteurs pour définir cette entité [2].
Du point de vue nosologique, trois sous-groupes de myosites à éosinophiles semblent se distinguer en fonction des caractéristiques cliniques et histologiques : une atteinte musculaire focale (la myosite focale à éosinophiles, MFE), et deux atteintes musculaires diffuses (la myosite diffuse à éosinophiles (MDE) et la périmyosite à éosinophiles (PE)) [3, 4].
L’étiologie des myosites à éosinophiles reste inconnue à ce jour. Certains auteurs ont soulevé l’hypothèse de facteurs déclenchants comme les traumatismes (notamment pour la forme focale) ou la consommation d’alcool [3, 4]. Il n’existe pas de consensus non plus concernant leur prise en charge. La littérature fait principalement référence à l’utilisation de thérapies immunosuppressives, les corticoïdes constituant généralement un traitement de première ligne.
La réalisation de cette revue de la littérature a pour objectif de regrouper et de synthétiser les connaissances sur les myosites à éosinophiles, afin de mieux appréhender cette entité clinico-pathologique rare, et de discuter les principaux diagnostics différentiels. Une requête dans MEDLINE a permis d’extraire 453 articles référencés par le mot-clé « myosite à éosinophiles ».
Au total, 69 cas de ont été répertoriés et analysés, dont 17 cas de myosites focales à éosinophiles, 36 cas de myosites diffuses à éosinophiles et 16 cas de périmyosites à éosinophiles. Les principales caractéristiques cliniques et biologiques de chacune d’entre elles sont rapportées ci-dessous.
Myosites focales à éosinophiles (MFE)
La myosite focale à éosinophiles (MFE) correspond à une atteinte musculaire impliquant de manière isolée un seul muscle ou un groupe de quelques muscles. Le diagnostic de myosite focale peut être posé grâce au focal myositis simple score (un score comprenant trois items, clinique, histologique et paraclinique avec électromyogramme ou IRM), lequel est considéré comme positif si ≥ 2 [5], associé à la présence d’éosinophiles dans la biopsie musculaire. Les patients présentant une MFE avaient un âge médian de 48 ans, avec un sex-ratio à 1. Le début de la maladie était plutôt aigu ou subaigu. La présentation clinique consistait typiquement en une masse ou un œdème musculaire parfois douloureux. La myosite touchait habituellement les membres (supérieurs ou inférieurs) mais n’entraînait généralement pas de faiblesse musculaire. Dans certains cas, le tableau pouvait mimer une thrombose veineuse profonde [9]. Les patients rapportés ne présentaient pas de manifestations systémiques par ailleurs. Sur le plan biologique, il était noté une éosinophilie sanguine et une élévation des enzymes musculaires dans la moitié des cas, sans syndrome inflammatoire associé. L’IRM permettait de conforter le diagnostic en montrant un hypersignal limité à un muscle ou un groupe musculaire sur les séquences pondérées T2 Fat-Sat [6–11]. L’analyse histologique retrouvait un infiltrat inflammatoire fait d’éosinophiles, parfois associée à quelques fibres en nécrose/régénération. L’évolution était bénigne dans la majorité des cas mais une corticothérapie de courte durée pouvait s’avérer utile pour traiter les symptômes et notamment la douleur.
Myosites diffuses à éosinophiles (MDE)
Cette forme associe une atteinte musculaire diffuse, souvent proximale et symétrique, et un infiltrat inflammatoire éosinophilique identifié sur la biopsie musculaire. Les patients avaient un âge médian de 49 ans, avec un sex-ratio à 1. Les symptômes apparaissaient généralement sur un mode subaigu ou chronique, et consistaient en des myalgies et/ou une faiblesse musculaire. Des signes extra-musculaires accompagnaient parfois le tableau : un phénomène de Raynaud ou des signes pulmonaires, neurologiques ou cardiologiques. Une éosinophilie sanguine était très fréquemment retrouvée, ainsi qu’une élévation des enzymes musculaires (CPK). L’IRM montrait souvent la présence de lésions inflammatoires au sein du muscle (hypersignal musculaire en séquence pondérée T2 ou T2-Fat-Sat), selon une distribution proximale et symétrique [12]. L’analyse histologique retrouvait des lésions de nécrose et de régénération des fibres musculaires associées à un important infiltrat inflammatoire fait d’éosinophiles allant jusqu’à l’endomysium [1–4, 13]. La majorité des patients ont bénéficié d’une corticothérapie permettant une résolution ou une amélioration des symptômes dans deux tiers des cas.
Périmyosites à éosinophiles (PE)
La périmyosite à éosinophiles se différencie de la MDE par un infiltrat inflammatoire musculaire fait d’éosinophiles ne dépassant pas le périmyisum, i.e. ne pénétrant pas entre les fibres musculaires [14]. Les patients avaient un âge médian de 44 ans, avec un sex ratio de 1,3 reflétant une légère prédominance masculine. Le début était, comme pour la MDE, subaigu ou chronique. La grande différence avec la MDE consiste en une atteinte musculaire subjective (myalgies) sans faiblesse musculaire. Dans le cadre de la PE, il a été rapporté une proportion importante de manifestations cutanées (induration, œdème et/ou urticaire) associées aux myalgies. L’éosinophilie sanguine était fréquemment retrouvée alors que les enzymes musculaires restaient normales. La majorité des patients ont bénéficié d’une corticothérapie permettant une résolution ou une amélioration des symptômes dans la moitié des cas.
Physiopathogénie
Les polynucléaires éosinophiles sont connus pour leur rôle dans les infections parasitaires ou immunoallergiques, mais également pour leur implication dans l’homéostasie tissulaire [15]. Leur production au sein de la moelle osseuse est stimulée, entre autres, par l’interleukine-5 (IL-5). Chaque éosinophile contient des granules cytotoxiques, libérés localement après activation ou trigger, pouvant conduire à des lésions des myofibres et à un dérèglement de l’homéostasie musculaire [16, 17]. L’implication de ce type de cellules dans les lésions musculaires pourrait expliquer la résistance au traitement observée chez certains patients.
Les principaux diagnostics différentiels
La fasciite à éosinophiles
La fasciite à éosinophiles (ou maladie de Schulman) est définie par une inflammation du fascia dont l’infiltrat inflammatoire polymorphe est associé à une éosinophilie sanguine. Les manifestations cutanées sont au premier plan : avec une induration sous-cutanée identifiée par le signe dit « du canyon », épargnant généralement les mains et le visage, ainsi que des œdèmes des membres [18, 19]. Des myalgies sont fréquemment rapportées par les patients, mais sans élévation concomitante des enzymes musculaires [19]. D’un point de vue radiologique, l’IRM retrouve un épaississement avec prise de contraste des fascias musculaires, sans signe d’atteinte musculaire ou osseuse. Sur le plan histologique, en plus de l’infiltrat inflammatoire polymorphe du fascia, des auteurs ont décrit chez certains patients une « myosite de continuité ». Peu documentées, ces lésions semblent plutôt périfasciculaires et potentiellement secondaires à la dégranulation locale des éosinophiles [19, 20]. L’ensemble de ces éléments en font un diagnostic différentiel relativement difficile à affirmer.
Les étiologies infectieuses
Les étiologies infectieuses, en particulier parasitaires, sont à éliminer, notamment chez un patient revenant d’une zone d’endémie pour Schistosoma, Trichinella, Toxocara, ou Sarcocystis [21]. Ces parasites peuvent être identifiés directement sur la biopsie musculaire.
Les étiologies génétiques
Deux pathologies musculaires héréditaires sont associées à la présence d’infiltrats d’éosinophiles intramusculaires. Il s’agit d’un diagnostic différentiel à évoquer tout particulièrement chez l’enfant ou l’adolescent. Des cas de myosites à éosinophiles secondaires à des mutations autosomiques récessives du gène CAPN3 (codant la calpaïne-3) ont été rapportés à plusieurs reprises [22, 23]. De tels infiltrats peuvent également exister dans la gamma-sarcoglycanopathie, une autre forme de dystrophie musculaire des ceintures [24]. Confronté à une myosite à éosinophiles, le clinicien se doit d’évoquer d’une cause génétique, notamment en cas : d’un âge de début jeune, d’une prédominance du déficit musculaire au niveau pelvien et scapulaire, d’une élévation persistante et très marquée des enzymes musculaires, d’une présence de lésions dystrophiques sur la biopsie musculaire (nécrose, régénération, fibrose, adipose) et d’une éventuelle cortico-résistance [25, 26].
Les étiologies auto-immunes
De très rares cas de granulomatose éosinophilique avec polyangéite peuvent mimer un tableau de ME [27, 28]. L’association à des manifestations extra-musculaires non cutanées, ainsi que la présence de granulomes sur la biopsie musculaire, peuvent aider à en établir le diagnostic.
Les étiologies toxiques
Le syndrome éosinophilie-myalgies, décrit initialement dans les années 1990, résulte de la consommation de L-tryptophane, un composé dont la toxicité musculaire est bien connue [28]. La présentation clinique peut être similaire à celle d’une ME, à savoir des myalgies sévères et diffuses associées à une éosinophilie sanguine. Le tableau clinique peut comprendre des manifestations neurologiques et cutanées [13, 29], lesquelles s’améliorent à l’arrêt de la consommation de L-tryptophane.
Le syndrome hyperéosinophilique
Dans certains cas, le syndrome hyperéosinophilique peut correspondre aux critères de ME du fait de sa définition (éosinophilie > 1,5 × 109/L associée à une atteinte d’organe). Cependant, les symptômes musculaires sont peu rapportés dans cette pathologie [30].
Conclusion
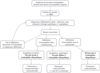
Les myosites à éosinophiles idiopathiques sont des entités rares associant des manifestations musculaires et avec une infiltration de polynucléaires éosinophiles dans le muscle. Le diagnostic repose en conséquence sur les données de la biopsie musculaire. Plusieurs formes sont décrites, allant d’une forme focale, bénigne à des formes diffuses et donc plus sévères nécessitant des traitements systémiques. La myosite diffuse à éosinophiles présente une atteinte musculaire objectivable cliniquement alors que la périmyosite à éosinophiles se présente avec des myalgies sans atteinte musculaire objectivable. Les principaux diagnostics différentiels sont la fasciite à éosinophiles, les myopathies héréditaires (calpaïnopathie ou autre) et les myosites infectieuses. Le traitement, en l’absence de consensus, repose sur l’utilisation de corticostéroïdes dans les formes diffuses. D’autres traitements ciblant plus spécifiquement la production de polynucléaires éosinophiles, comme les anti-IL5, pourraient se discuter dans les cas avec atteinte réfractaire et éosinophilie sanguine importante [31] (Figure 1).
Liens d’intérêt
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Références
- Layzer RB, Shearn MA, Satya-Murti S. Eosinophilic polymyositis. Ann Neurol 1977 ; 1 : 65–71. [CrossRef] [PubMed] [Google Scholar]
- Kumamoto T, Ueyama H, Fujimoto S, et al. Clinicopathologic characteristics of polymyositis patients with numerous tissue eosinophils. Acta Neurol Scand 1996 ; 94 : 110–4. [CrossRef] [PubMed] [Google Scholar]
- Selva-O’Callaghan A, Trallero-Araguás E, Grau JM. Eosinophilic myositis: an updated review. Autoimmun Rev 2014 ; 13 : 375–8. [CrossRef] [PubMed] [Google Scholar]
- Hall FC, Krausz T, Walport MJ. Idiopathic eosinophilic myositis. QJM Mon J Assoc Physicians 1995 ; 88 (8) : 581–6. [Google Scholar]
- Gallay L, Hot A, Petiot P, et al. Focal myositis: New insights on diagnosis and pathology. Neurology 2018 ; 90 : e1013–20. [CrossRef] [PubMed] [Google Scholar]
- Muqit MMK, Taylor RH. Cyclical orbital eosinophilic myositis. Br J Ophthalmol 2007 ; 91 : 1569–70. [CrossRef] [PubMed] [Google Scholar]
- Galindo-Ferreiro A, Torres Nieto MA, Barbado Ajo J, et al. Focal eosinophilic myositis affecting the orbicularis muscle: Clinical and image aspects. Ophthal Plast Reconstr Surg 2020 ; 36 : e51–3. [CrossRef] [PubMed] [Google Scholar]
- Roh YH, Koh YD, Noh JH, et al. Focal Eosinophilic myositis of the hand presenting as a refractory pyogenic infection. J Hand Surg 2017 ; 42 (842) : e1–842.e3. [CrossRef] [Google Scholar]
- Sharma P, Sharma MC. Resolving Eosinophilic Myositis: MR Features. J Comput Assist Tomogr 2001 ; 25 : 151–3. [CrossRef] [PubMed] [Google Scholar]
- Hamada S, Ueki S, Miyabe Y, et al. Focal eosinophilic myositis with Charcot-Leyden crystal formation. Allergol Int Off J Jpn Soc Allergol 2020 ; 69 : 633–5. [CrossRef] [Google Scholar]
- Yamashita S, Kawasumi H, Kimura M, et al. Spontaneous resolution of focal eosinophilic myositis of the adductor pollicis complicated by lung lesions. Mod Rheumatol Case Rep 2020 : 106–9. [CrossRef] [PubMed] [Google Scholar]
- Nakashima M, Kawabe Y, Aoyagi T, et al. A case of eosinophilic myositis associated with orbital myositis. Mod Rheumatol 2002 ; 12 : 80–3. [CrossRef] [PubMed] [Google Scholar]
- Kaufman LD, Kephart GM, Seidman RJ, et al. The spectrum of eosinophilic myositis. Clinical and immunopathogenic studies of three patients, and review of the literature. Arthritis Rheum 1993 ; 36 : 1014–24. [CrossRef] [PubMed] [Google Scholar]
- Serratrice G, Pellissier JF, Roux H, et al. Fasciitis, perimyositis, myositis, polymyositis, and eosinophilia. Muscle Nerve 1990 ; 13 : 385–95. [CrossRef] [PubMed] [Google Scholar]
- Kita H, Eosinophils : multifaceted biological properties and roles in health and disease. Immunol Rev 2011 ; 242 : 161–77. [CrossRef] [PubMed] [Google Scholar]
- Cantarini L, Volpi N, Carbotti P, et al. Eosinophilia-associated muscle disorders: an immunohistological study with tissue localisation of major basic protein in distinct clinicopathological forms. J Clin Pathol 2009 ; 62 : 442–7. [CrossRef] [PubMed] [Google Scholar]
- Sugihara R, Kumamoto T, Ito T, et al. Human muscle protein degradation in vitro by eosinophil cationic protein (ECP). Muscle Nerve 2001 ; 24 : 1627–34. [CrossRef] [PubMed] [Google Scholar]
- Shulman LE. Diffuse fasciitis with eosinophilia: a new syndrome? Trans Assoc Am Physicians 1975 ; 88 : 70–86. [PubMed] [Google Scholar]
- Lebeaux D, Sène D. Eosinophilic fasciitis (Shulman disease). Best Pract Res Clin Rheumatol 2012 ; 26 : 449–58. [CrossRef] [PubMed] [Google Scholar]
- Toquet C, Hamidou MA, Renaudin K, et al. In situ immunophenotype of the inflammatory infiltrate in eosinophilic fasciitis. J Rheumatol 2003 ; 30 : 1811–5. [PubMed] [Google Scholar]
- El-Beshbishi SN, Ahmed NN, Mostafa SH, et al. Parasitic infections and myositis. Parasitol Res 2012 ; 110 : 1–18. [CrossRef] [PubMed] [Google Scholar]
- Krahn M, Goicoechea M, Hanisch F, et al. Eosinophilic infiltration related to CAPN3 mutations: a pathophysiological component of primary calpainopathy? Clin Genet 2011 ; 80 : 398–402. [CrossRef] [PubMed] [Google Scholar]
- Krahn M, Hanisch F, Goicoechea M, et al. CAPN3 mutations in patients with idiopathic eosinophilic myositis: A predystrophic stage of LGMD2A? Neuromuscul Disord 2007 ; 17 : 791–2. [CrossRef] [Google Scholar]
- Baumeister SK, Todorovic S, Milić-Rasić V, et al. Eosinophilic myositis as presenting symptom in gamma-sarcoglycanopathy. Neuromuscul Disord 2009 ; 19 : 167–71. [CrossRef] [PubMed] [Google Scholar]
- Brown RH, Amato A. Calpainopathy and eosinophilic myositis. Ann Neurol 2006 ; 59 : 875–7. [CrossRef] [PubMed] [Google Scholar]
- Schröder T, Fuchss J, Schneider I, et al. Eosinophils in hereditary and inflammatory myopathies. Acta Myol 2013 ; 32 : 148–53. [PubMed] [Google Scholar]
- Parent ME, Larue S, Ellezam B. Eosinophilic granulomatosis with polyangiitis (Churg-Strauss syndrome) presenting as diffuse myositis. BMC Musculoskelet Disord 2014 ; 15 : 388. [CrossRef] [PubMed] [Google Scholar]
- Uehara M, Hashimoto T, Sasahara E, et al. Churg-Strauss syndrome presenting as myositis following unaccustomed exercise. J Clin Neurosci 2009 ; 16 : 1232–3. [CrossRef] [PubMed] [Google Scholar]
- Tamaki H, Chatterjee S, Langford CA. Eosinophilia in Rheumatologic/Vascular Disorders. Immunol Allergy Clin North Am 2015 ; 3 : 453–76. [CrossRef] [PubMed] [Google Scholar]
- Ogbogu PU, Bochner BS, Butterfield JH, et al. Hypereosinophilic syndrome: A multicenter, retrospective analysis of clinical characteristics and response to therapy. J Allergy Clin Immunol 2009 ; 124 : 1319–25. [CrossRef] [PubMed] [Google Scholar]
- Waheed S, Zubair H, Waheed F. Treatment of eosinophilic myositis with mepolizumab. Indian J Rheumatol 2018 ; 13 : 289. [CrossRef] [Google Scholar]
NOTE
Cet article est largement inspiré de travaux publiés dans la revue Neuromuscular Disorders. (Fermon C, Authier FJ, Gallay L. Neuromuscul Disord 2022; 32 :116-124).
Liste des figures
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.