| Numéro |
Cah. Myol.
Numéro 17, Juin 2018
|
|
|---|---|---|
| Page(s) | 15 - 18 | |
| Section | Cas cliniques / Case reports | |
| DOI | https://doi.org/10.1051/myolog/201817004 | |
| Publié en ligne | 6 juin 2018 | |
Un cas de myopathie myofibrillaire infantile dû à une mutation dans le gène FLNC
Infantile onset myofibrillar myopathy as a new phenotype of Filamin C mutation
1
Laboratoire de Cardiogénétique Moléculaire, Centre de Biologie et Pathologie Est, Bron, France
2
Service d’Épileptologie Clinique, des Troubles du Sommeil et de Neurologie Fonctionnelle de l’Enfant. Hôpital Femme Mère Enfant, Hospices Civils de Lyon, France
3
Institut NeuroMyoGène, CNRS UMR 5310, Inserm U1217, Université Lyon 1, Lyon, France
4
Service de Neuropathologie, Centre de Biologie et Pathologie Est, Hospices Civils de Lyon, France
* Contact alexandre.janin01@chu-lyon.fr
Les myopathies myofibrillaires sont un groupe de pathologies cliniquement et génétiquement hétérogènes mais partageant des caractéristiques histologiques communes. On retrouve au niveau du muscle des modifications de la structure des myofibrilles associées à une accumulation intracellulaire de protéines. Les manifestations cliniques sont variables d’un individu à l’autre mais marquées par une faiblesse musculaire généralement lentement progressive. À l’heure actuelle, neuf gènes codant des protéines faisant partie de la strie Z ont été identifiés à ce jour comme responsables de myopathie fibrillaire.
© A. Janin et al., publié par EDP Sciences, 2018
 Cet article est distribué sous licence « Creative Commons » : http://creativecommons.org/licenses/by/4.0/deed.fr/, permettant une ré-utilisation du contenu sans restriction à condition de mentionner clairement la source.
Cet article est distribué sous licence « Creative Commons » : http://creativecommons.org/licenses/by/4.0/deed.fr/, permettant une ré-utilisation du contenu sans restriction à condition de mentionner clairement la source.
Observation
L’enfant F, née en 2010, est la cadette d’une fratrie de quatre enfants issus de parents cousins germains. Elle est née à 37 semaines d’aménorrhée d’une grossesse de déroulement normal.
À la naissance, la jeune F a présenté une hypotonie congénitale. Au cours du premier mois de vie, des troubles de la succion et de la déglutition ont été mis en évidence.
Elle a développé, à 10 mois, une déformation du rachis cervical. Elle acquiert la marche à l’âge de 18 mois avec un pied droit en rotation interne. Progressivement, sont apparues des malformations des articulations associées à un cou court et raide. Une scoliose thoraco-lombaire maligne s’installe progressivement combinée à une rigidité des ceintures pelvienne et scapulaire. Elle présente également une déformation asymétrique des pieds avec un varus adductus de l’avant pied droit. Une atrophie musculaire au niveau du membre inférieur (cuisse et mollet) associée à une hyporéflexie ont été mises en évidence. Elle peut lever les bras à l’horizontale mais pour une durée limitée, témoin d’une faiblesse musculaire proximale. La petite F se plaint également de difficultés à écrire. Le passage de la position assise à la position debout nécessite l’aide de ses mains avec un signe de Gowers positif. Son temps de marche est réduit et inférieur à dix minutes. L’atteinte faciale se limite à une asymétrie palpé- brale associée à une légère inclinaison de sa tête sur la gauche. Elle présente également des apnées du sommeil nécessitant une ventilation nocturne non invasive. Une cardiomyopathie hypertrophique peu symptomatique, associée à une communication interventriculaire trabéculée apicale de type I, ont été détectées. L’absence de troubles du rythme associé est objectivée par un holter ECG à l’occasion d’un épisode de palpitations ( Figure 1).
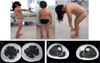 |
Figure 1 Photographies (A-C) et IRM des cuisses (D) et des mollets (E) de lajeune F témoignant de l’atteinte musculaire. |
L’exploration paraclinique a mis en évidence une activité sérique de la créatine kinase élevée à 473 UI/L (valeurs usuelles < 170 UI/L). Les IRM médullaire et cérébrale sont normales. L’IRM musculaire montre une infiltration adipeuse prédominante au niveau des membres inférieurs avec un aspect laminé, surtout au niveau des mollets.
Une biopsie musculaire a été effectuée dans le quadriceps gauche. Elle montrait des fibres de type I et II hypertrophiées avec des corps intra-myofibrillaires et une prédominance de fibres de type I associée à de nombreuses fibres de type II atrophiques révélées par la réaction ATPase 4.63. La coloration PAS confirmait la composition non glycogénique de ces agrégats et la coloration à la phosphatase acide montrait une augmentation de l’activité lysosomale autour de ces corps. Aucune spécificité de fibres de ces agrégats n’était retrouvée ( Figure 2). Les immunohistochimies standard et l’étude en Western-blot des protéines habituellement impliquées dans les dystrophies musculaires sont normales. L’étude en microscopie électronique n’a pas permis de mettre en évidence de dépôt caractérisitique. L’expression du collagène 6 dans le muscle et dans les fibroblastes cutanés est normale. L’étude de la glycogénolyse et de la glycolyse ne montrent pas d’anomalies. Enfin, l’aspect réducteur des agrégats au trichrome de Gomori a d’abord orienté le diagnostic vers une myopathie à corps réducteurs. Dans le but de mettre en évidence une cause moléculaire à ce premier diagnostic de myopathie à corps réducteurs, un séquençage ciblé par la méthode de Sanger des gènes FHL1, RYR1 et LMNA a été effectué. Cependant, aucune variation pathogène ou probablement pathogène n’a pu être mise en évidence. Le marquage à la ménadione effectué afin d’objectiver le caractère réducteur des corps identifiés s’est révélé négatif. Le diagnostic de « myopathie à corps réducteurs » a alors été remis en cause et celui de « myopathie myofibrillaire » a été retenu ( Figure 2).
 |
Figure 2 Examen histologique de la biopsie musculaire. La coloration HES (A) et le trichrome de Gomori (B) montrentd’importants agrégats réducteurs dans lesfibres. (C) La colorationPAStémoigne de leur compositionnon glycogénique. (D)Lacoloration par la phosphatase acide montre une activité lysosomale augmentée autour des agrégats. (E) L’ATPase 4.63 montre que les agrégats sont présents autant dans les deux types de fibres. (F) Le marquage à la ménadione montre que ces agrégats ne sont pas réducteurs, ce qui est en faveur du diagnostic de myopathie myofibrillaire. |
Ainsi, afin de compléter le bilan génétique et parce que la jeune F souffre désormais d’une cardiomyopathie hypertrophique, une exploration moléculaire basée sur l’étude de 48 gènes impliqués dans les cardiomyopathies par séquençage à haut débit a été effectuée [1]. Parmi les variants génomiques identifiés, seule une variation faux-sens présente à l’état hétérozygote dans le gène FLNC, codant la filamine C, pouvait être considérée comme variant probablement pathogène. Selon les recommandations de l’American Collège of Médical Genetics [2], aucun autre variant pathogène ou probablement pathogène n’a été identifié, bien que la consanguinité parentale nous orientait au départ plutôt vers une pathologie autosomique récessive. Ce variant (p.Gly1168Asp, NM_001458.4:c.3503G>A), confirmé par séquençage selon la méthode de Sanger, aboutit à un changement d’acide aminé dans le dixième domaine immunoglobulin-like de la protéine. Iln’apas été rapporté (mars 2018) dans la base de données « contrôles » telles que GnomAD. Il n’a également pas été identifié au sein de notre cohorte de 721 patients souffrant de cardiomyopathies hypertrophiques et étudiés avec le même panel de gènes. L’acide aminé concerné est très conservé à travers les espèces. Les différents algorithmes in silico utilisés (AlignGVGD, PolyPhen-2, Mutation Taster, SIFT et UMD-predictor) pour mieux évaluer le caractère pathogène de cette nouvelle variation concluent tous à un effet délétère sur la fonction de la protéine. Dans le but de mieux appréhender son éventuel caractère pathogène, une étude de ségrégation dans la famille a été effectuée. La présence de la variation a été testée chez tous les membres de la famille ayant accepté l’exploration. Aucun des parents du cas index, qui sont tous les deux sains, ne porte la variation. Ceci suggère que la variation identifiée chez la jeune F est une variation de novo.
Un immunomarquage de la filamine C a été effectué sur des coupes transversales de quadriceps pour conclure sur la nature des agrégats observés dans les fibres musculaires. Cette expérience a permis de mettre en évidence des agrégats de filamine C dans les fibres musculaires du cas index, agrégats absents de coupes transversales effectuées chez un témoin sain ( Figure 3).
 |
Figure 3 L’immunofluorescence montre de larges agrégats de filamine C dans les fibres musculaires. |
Commentaire
Le concept de « myopathie myofibrillaire » (MFM) a été proposé en 1996 par A. Engel comme un groupe de pathologies partageant des caractéristiques histologiques communes telles que l’accumulation intra-sarcoplasmique de protéines, l’expression ectopique de protéines (dont la plupart sont des constituants de la strie Z), la présence de vacuoles ou encore la désorganisation du réseau entre les myofibrilles [3]. Jusqu’à présent, neuf gènes principaux sont classiquement responsables de MFM : DES, CRYAB, LDB3/ZASP, MYOT, BAG3, FHL1, TTN, ACTA1 et FLNC.
Comme le montre cette observation, et conformément à la littérature, le diagnostic repose sur la biopsie musculaire. Les caractéristiques histologi- ques, quelle que soit la cause moléculaire, regroupent la présence de dépôts granuleux ou amorphes de taille et contour variables au trichrome, des foyers avec une réduction voire une perte d’activité enzymatique oxydante, la présence de vacuoles, des anomalies compatibles avec une origine neurogène telles que des groupes de fibres atrophiques et enfin l’expression ectopique de protéines dont la plupart sont normalement associées aux stries Z (desmine, aB-cristalline, myotiline ou filamine C). La microscopie électronique est également une aide précieuse pour le diagnostic car elle permet de mettre en évidence la désorganisation myofibrillaire et l’accumulation focale de protéines dégradées. L’ensemble de ces anomalies histologiques, bien que non spécifiques, permet de poser le diagnostic de MFM [4]. Cependant, les anomalies associées aux MFM étant focales, une première biopsie peut s’avérer négative ou peu contributive, et une seconde peut alors être nécessaire.
Comme le montre ce cas, des études immunohisto- chimiques sont des outils sensibles permettant de mettre en évidence l’agrégation protéique sur la biopsie musculaire [4].
Le premier cas de MFM due à une mutation faux- sens dans le gène FLNC a été rapporté en 2005 [5]. Habituellement, les MFM par mutation dans le gène codant la filamine C sont associées au départ à une faiblesse musculaire touchant surtout les muscles proximaux et débutant entre la quatrième et la cinquième décade [6, 7]. Ensuite, la plupart des patients développent une faiblesse musculaire des muscles distaux et proximaux des membres supérieurs et inférieurs pouvant conduire à la perte de la marche. Une atteinte des muscles respiratoires, nécessitant une ventilation nocturne, est également retrouvée dans l’évolution de la pathologie. Une atteinte cardiaque, de type troubles de la conduction, hypertrophie du ventricule gauche ou troubles diastoliques, est associée chez environ 30% des patients [7]. Sur le plan paraclinique, une augmentation de l’activité créatinine kinase jusqu’à 10 fois la valeur usuelle supérieure peut être retrouvée.
Nous rapportons ici, à notre connaissance, le premier cas de myopathie myofibrillaire débutant dès la petite enfance et associée à une mutation dans le gène FLNC. Ce cas illustre d’abord l’intérêt d’une collaboration étroite entre les équipes du service clinique, du laboratoire d’anatomopathologie et du laboratoire de biologie moléculaire pour le diagnostic des pathologies musculaires. Il montre également une nouvelle fois l’utilité du séquençage à haut débit pour explorer rapidement un nombre important de gènes de manière rapide et augmenter ainsi le nombre de patients avec un diagnostic moléculaire positif.
Liens d’intérêt
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Remerciements
Les auteurs souhaitent remercier le Dr Carole Chevenet (CHU Clermont-Ferrand) pour son aide pour la réalisation du marquage à la ménadione ainsi que Tissu Tumorotech Est.
Références
- Chanavat V, Janin A, Millat G. A fast and cost-effective molecular diagnostic tool for genetic diseases involved in sudden cardiac death. Clin Chim Acta 2016; 453: 80–5. [CrossRef] [Google Scholar]
- Richards S, Aziz N, Bale S, et al. ACMG Laboratory quality assurance committee, standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med 2015; 17: 405–24. [CrossRef] [PubMed] [Google Scholar]
- Nakano S, Engel AG, Waclawik AJ, et al. Myofibrillar myopathy with abnormal foci of desmin positivity. I. Light and electron microscopy analysis of 10 cases. J Neuropathol Exp Neurol 1996; 55: 549–62. [CrossRef] [PubMed] [Google Scholar]
- Schröder R, Schoser B. Myofibrillar myopathies: a clinical and myopathological guide. Brain Pathol 2009; 19: 483–92. [CrossRef] [PubMed] [Google Scholar]
- Vorgerd M, van der Ven PFM, Bruchertseifer V, et al. A Mutation in the dimerization domain of filamin C causes a novel type of autosomal dominant myofibrillar myopathy. Am J Hum Genet 2005; 77: 297–304. [CrossRef] [Google Scholar]
- Kley RA, Hellenbroich Y, van der Ven PFM, et al. Clinical and morphological phenotype of the filamin myopathy: a study of 31 German patients. Brain J Neurol 2007; 130: 3250–64. [CrossRef] [PubMed] [Google Scholar]
- Fürst DO, Goldfarb LG, Kley RA, et al. Filamin C-related myopathies: pathology and mechanisms. Acta Neuropathol 2013; 125: 33–46. [CrossRef] [PubMed] [Google Scholar]
Liste des figures
|
Figure 1 Photographies (A-C) et IRM des cuisses (D) et des mollets (E) de lajeune F témoignant de l’atteinte musculaire. |
| Dans le texte | |
|
Figure 2 Examen histologique de la biopsie musculaire. La coloration HES (A) et le trichrome de Gomori (B) montrentd’importants agrégats réducteurs dans lesfibres. (C) La colorationPAStémoigne de leur compositionnon glycogénique. (D)Lacoloration par la phosphatase acide montre une activité lysosomale augmentée autour des agrégats. (E) L’ATPase 4.63 montre que les agrégats sont présents autant dans les deux types de fibres. (F) Le marquage à la ménadione montre que ces agrégats ne sont pas réducteurs, ce qui est en faveur du diagnostic de myopathie myofibrillaire. |
| Dans le texte | |
|
Figure 3 L’immunofluorescence montre de larges agrégats de filamine C dans les fibres musculaires. |
| Dans le texte | |
Les statistiques affichées correspondent au cumul d'une part des vues des résumés de l'article et d'autre part des vues et téléchargements de l'article plein-texte (PDF, Full-HTML, ePub... selon les formats disponibles) sur la platefome Vision4Press.
Les statistiques sont disponibles avec un délai de 48 à 96 heures et sont mises à jour quotidiennement en semaine.
Le chargement des statistiques peut être long.