| Issue |
Cah. Myol.
Number 13, Juin 2016
|
|
|---|---|---|
| Page(s) | 15 - 24 | |
| Section | Prise en charge / Management | |
| DOI | https://doi.org/10.1051/myolog/201613005 | |
| Published online | 11 juillet 2016 | |
eDystrophin : un nouvel outil dédié à une meilleure compréhension des dystrophinopathies
eDystrophin: a new tool for better understanding dystrophinopathies
1
Centre de Recherche en Myologie ; Inserm UMRS 974 ; CNRS FRE 3617 ; Cellule bases de données ; Institut de Myologie, 75651 Paris, France
2
APHP, Groupe Hospitalier Universitaire la Pitié-Salpêtrière, Centre de Référence de Pathologie Neuromusculaire Paris-Est, Institut de Myologie, 75651 Paris, France
3
Université de Rennes 1, Avenue du Professeur Léon Bernard, 35043 Rennes, France
4
CNRS UMR 6290, Institut de Génétique et Développement de Rennes, 35043 Rennes, France
5
APHP, Hôpital Cochin, laboratoire de biochimie et génétique moléculaire, 75014 Paris, France
* Contact Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Les dystrophies musculaires de Duchenne (DMD) et de Becker (BMD) sont deux formes phénotypiques sous-tendues par l’atteinte d’un même gène : le gène DMD. Classiquement, le phénotype sévère DMD résulte d’une mutation empêchant toute production de la protéine correspondante, la dystrophine, alors que le phénotype atténué BMD résulte de mutations compatibles avec la synthèse d’une dystrophine anormale en quantité et/ou en qualité.
La dystrophine principale du muscle (isoforme dp427m) est une protéine filamenteuse de type spectrine où l’on distingue quatre grands domaines de tailles différentes [14, 16] (Figure 1) (domaines N-terminal, central, riche en cystéine et C-terminal) (Figure 1). Chacun de ces domaines interagit avec plusieurs partenaires de la cellule musculaire dont le nombre va croissant et dont l’identification est loin d’être achevée. Leur connaissance est essentielle pour élucider la fonctionnalité de la dystrophine normale et de ses variants pathologiques. Le domaine central en particulier possède de multiples interactions avec les lipides membranaires qui maintiennent la dystrophine en contact avec le sarcolemme [17, 18], l’actine [2], la sérine/thréonine kinase Par-1b [39], les filaments intermédiaires de type synémines α et β [4], les microtubules [29] et une protéine enzymatique jouant un rôle clé dans l’activité musculaire, l’oxyde nitrique synthase neuronale ou nNOS [15, 20].
 |
Figure 1. Les différents grands domaines de la dystrophine et les protéines interagissant avec elle. Voir texte pour les abréviations. |
Les premières mutations du gène DMD ont été identifiées il y a 30 ans. La connaissance du spectre mutationnel de ce gène a progressivement permis de mieux cerner les corrélations entre le génotypeDMD et le phénotype résultant. La grande majorité de ces mutations sont des délétions intragéniques entraînant la perte d’un ou plusieurs exons [1, 19, 34]. Les duplications exoniques et les mutations ponctuelles de divers types représentent aussi une part non négligeable des événements mutationnels du gène DMD. Comme le veut la règle de Monaco [21], lorsque le cadre de lecture résiduel est aboli, aucune protéine n’est produite et le phénotype est de type DMD, lorsqu’il est maintenu, une dystrophine tronquée est produite avec un phénotype BMD. Cette règle permet d’expliquer la différence phénotypique entres les deux formes alléliquesDMDet BMD. C’est sur ce principe que les stratégies thérapeutiques de saut d’exon au niveau transcriptionnel sont fondées [35]. Encore faut-il que la dystrophine tronquée ainsi produite après rétablissement du cadre de lecture ait une fonctionnalité suffisante permettant par exemple de « transformer » un malade DMD en un BMD. Or, comme l’indiquent les cas de phénotypes sévères, BMDsévères voire DMD, résultant de délétions théoriquement en phase, il est possible que la dystrophine tronquée résiduelle soit de qualité médiocre. Cela met en exergue non seulement l’importance de la production d’une dystrophine « viable » mais aussi de bonne qualité. Ainsi, toutes les dystrophines issues de mutations non-tronquantes (appelées aussi « quasidystrophines ») ne se valent pas [16]. Les travaux de Chamberlain sur les micro-dystrophines et mini-dystrophines ont permis de préciser les exons nécessaires et les exons indispensables permettant de maintenir une dystrophine fonctionnelle [12]. La perte d’interaction avec ses partenaires, l’effet « modificateur » de variants sur d’autres gènes et le retentissement sur la configuration secondaire et tertiaire de la quasi-dystrophine issues de mutations non-tronquantes sont autant de pistes prometteuses qui permettront non seulement de comprendre la variabilité phénotypique au sein des dystrophinopathies [11, 26] mais aussi de sélectionner les quasidystrophines les plus optimales dans le cadre du saut d’exon thérapeutique.
Dans cette optique, nous avons développé eDystrophin, une banque de données librement accessible sur internet et interactive permettant non seulement de prédire les conséquences structurales des mutations du gène DMD conservant le cadre de lecture mais aussi leurs conséquences phénotypiques et protéiques observées chez les patients qui en sont porteurs.
La dystrophine : le gène et la protéine
La dystrophine est une protéine encodée par un énorme gène, appelé DMD 1, situé sur le bras court du chromosome X en Xp21.2 [13, 22]. Il comporte 2,4 millions de paires de bases, ce qui en fait le plus grand gène humain, et est composé de 79 exons. Les introns représentent 99 % de la taille du gène. Ce gène permet la synthèse de 7 isoformes principales de la dystrophine, de tailles différentes, grâce à la présence de 7 promoteurs tissus-spécifiques. Trois de ces promoteurs (appelés promoteurs M, B et P) sont situés en amont du premier exon et permettent la synthèse des dystrophines pleine longueur fonctionnellement équivalentes car ne diffèrent que par les quelques premiers acides aminés de leurs extrémités N-terminales (dp427m, dp427b et dp427p) alors que les 4 autres promoteurs sont intragéniques et permettent la synthèse d’isoformes de taille réduite (dp260, dp140, dp116, dp71). Nous ne nous étendrons pas sur les nombreuses isoformes secondaires produites par épissages alternatifs internes au gène. La dystrophine pleine longueur est constituée de 3 685 acides aminés pour un poids moléculaire de 427 kDa. Elle est présente dans les tissus musculaires squelettique et cardiaque, le cerveau, la rétine, les cellules gliales et de Purkinje et en quantité très réduite dans les lymphocytes. Elle fait partie de la grande famille de protéines filamenteuses de type « spectrine », caractérisées par un domaine N-terminal se liant à l’actine ainsi que par la présence de motifs répétés homologues à ceux de la spectrine appelés « répétitions ».
La dystrophine est composée de quatre grands domaines [14, 16] (Figure 1 ) :
-
Le domaine N-terminal ou domaine de liaison à l’actine (Actin Binding Domain ou ABD1), comprenant les premiers 246 acides aminés. Ce domaine est lui-même constitué de deux sous-domaines dits « homologues à la calponine » (CH1 et CH2) réunis par un fragment en hélice α. En plus de sa liaison à l’actine, ce domaine lie également les kératines 19 et 8.
-
Le domaine central (ou rod domain) va de l’acide aminé 247 au 3045. Il représente ainsi 76 % de la protéine. Il est constitué de 24 répétitions homologues à la spectrine (ou spectrine like, R1 à R24) et de 4 charnières riches en proline (ou « hinges », H1 à H4) divisant le domaine central en 3 sousdomaines. Ce domaine central possède diverses interactions avec les lipides membranaires via 2 domaines de liaison aux lipides (lipid binding domain ou LBD) au niveau des répétitions R1-R3 (LBD1) et R4-R19 (LBD2) qui maintiennent la dystrophine en contact avec le sarcolemme [17, 18], l’actine (domaine ABD2, répétitions 11-17) [2], la sérine/thréonine kinase Par-1b (répétitions R8-R9) [39], les synémines α et β qui sont des protéines de la famille des filaments intermédiaires (répétitions R11-R14) [4] et l’oxyde nitrique synthétase neuronale ou nNOS (répétitions R16-R17) [15, 20] et enfin les microtubules (répétitions R20-24) [29] permettant de maintenir près du sarcolemme cette protéine enzymatique jouant un rôle clé dans l’activité musculaire.
-
Le domaine riche en cystéine (cysteine rich repeats ou CRR) va de l’acide aminé 3080 à 3360. Il est composé de quatre sous-domaines : 1 domaine appelé « WW » (2 tryptophanes séparées par 21 acides aminés), 2 domaines en « main EF » (« EF hand », EFH1 et EFH2) organisés en hélice-bouclehélice et d’un domaine en doigt de zinc (ZZ). Le domaine CRR se lie avec le β-dystroglycane (β-DG) qui, en interagissant avec l’α-dystroglycane et les sarcoglycanes, forme le complexe dystrophine-glycoprotéines (DGC) [5, 9]. Il se lie aussi à la synémine α [4], la plectine [31], la myosprine (protéine kinase de type A) [29] et l’ankyrine [3].
-
Le domaine C-terminal formé des 325 derniers résidus est composé de faisceaux d’hélices similaires à ceux présents dans le domaine central. Il interagit avec la dystrobrévine, les syntrophines et la myosprine [24, 30, 32, 33].
La dystrophine est localisée sous la face interne du sarcolemme en lien intime avec les glycoprotéines sarcolemmiques formant le complexe DGC. Elle joue un rôle essentiel durant le cycle contractionrelaxation musculaire. En formant un lien entre la matrice extracellulaire et le cytosquelette d’actine, la dystrophine protège le sarcolemme de la rupture durant le stress induit par la contraction musculaire [27]. Plusieurs hypothèses ont été émises pour expliquer les mécanismes moléculaires mis en jeu. Ces hypothèses font référence à sa structure filamenteuse et à ses nombreux domaines de liaison.
Les dystrophinopathies : spectre clinique et mutationnel
Les dystrophinopathies sont l’ensemble des affections causées par des mutations du gène codant la dystrophine [6, 19, 23, 34]. Elles sont transmises selon le mode récessif lié à l’X. Nous ne nous étendrons pas ici sur les aspects cliniques détaillés. Les dystrophinopathies sont essentiellement représentées par :
- La DMD, dystrophie musculaire sévère de l’enfant, affectant 1 sur 3 500 à 6 000 garçons naissant par an, débutant classiquement vers 3-4 ans par des difficultés motrices aux membres inférieurs avec pseudohypertrophie des mollets, s’accentuant progressivement jusqu’à la perte de la marche autour de l’âge de 10 ans, avant 13 ans dans la grande majorité des cas. Apparaissent ensuite des atteintes musculaire axiale (cyspho-scoliose), respiratoire (insuffisance respiratoire restrictive) et cardiaque (cardiomyopathie dilatée) évolutives qui font toute la gravité de la maladie et sont souvent responsables du décès en général au cours de la 3e décennie.
La BMD est une dystrophie musculaire moins sévère que la DMD, de début plus tardif à l’adolescence ou l’âge adulte, avec difficultés motrices et hypertrophie musculaire, mais moins évolutive avec quand même impotence fonctionnelle pouvant être majeure et aller jusqu’à la perte de la marche, entre 40 et 60 ans. En revanche une atteinte cardiaque est constante chez les BMD, devenant patente à partir de la 4e décennie de vie, de sévérité variable pouvant nécessiter parfois une transplantation cardiaque. L’atteinte respiratoire est aussi présente mais beaucoup moins sévère et plus tardive que dans la DMD. Une atteinte cognitive de sévérité variable est plus fréquemment observée dans les DMD que dans les BMD, essentiellement retrouvée chez les patients porteurs de mutation en aval de l’exon 51.
La DMD et la BMD apparaissent ainsi comme les deux extrêmes du spectre phénotypique des dystrophinopathies. Entre ces deux entités, se retrouvent un certain nombre de patients dits « intermédiaires », perdant souvent la marche après 13 ans et avant 16 ans [8]. Exceptionnellement, certains patients, porteurs de certaines mutations, développent une cardiomyopathie dilatée sans atteinte musculaire squelettique associée avec cependant une élévation des CPK. À l’extrême bout du spectre clinique des dystrophinopathies, un certain nombre de patients a été rapporté sous l’étiquette de dystrophinopathies « pseudométaboliques » avec intolérance à l’effort, rhabdomyolyse et hypertrophie musculaire ou élévation isolée des CPK. Beaucoup de ces patients développent par la suite une symptomatologie musculaire déficitaire et une atteinte cardiorespiratoire rejoignant ainsi le phénotype BMD. Enfin, bien qu’affectant théoriquement les garçons, certaines femmes porteuses hétérozygotes pour la mutation DMD peuvent manifester des symptômes en cas de translocation équilibrée impliquant le chromosome X ou d’inactivation réciproque non équilibrée des chromosomes X. Dans ces cas, peut être observée une atteinte cardiaque isolée ou musculo-cardiaque ressemblant souvent à celle observée dans laBMDet plus rarement de laDMD(on parle alors de « Duchenne féminin »).
Les mutations du gène DMD responsables des dystrophinopathies sont représentées par des grandes délétions d’un ou plusieurs exons (60 à 70 % des cas), des mutations ponctuelles (i.e. petites insertions ou délétions d’un ou plusieurs nucléotides, non-sens (stops directs), introniques et faux-sens (15 à 20 %) et des grandes duplications d’un ou plusieurs exons (10 à 15 %) [1, 19, 34]. La région centrale du gène s’étendant entre les exons 45 et 55 est la plus pourvoyeuse de délétions exoniques avec très souvent des délétions débutant à l’exon 45 (point de cassure proximal dans l’intron 44).
Les corrélations phénotype/génotype au sein des dystrophinopathies
La variabilité phénotypique inter- et intrafamiliale des affections génétiques est largement connue. Comment expliquer que des mutations dans un même gène puissent être responsables d’expressions phénotypiques aussi différentes allant des patients pauci-symptomatiques à des affections aussi sévères que la DMD ?
1. La règle de Monaco et ses exceptions
La référence pour expliquer la différence phénotypique entres les deux formes alléliques DMD et BMD est la règle proposée par Tony Monaco [21] sur la conservation ou non d’un cadre de lecture ouvert du transcrit (l’ARN) muté de la dystrophine. Si la mutation a pour conséquence d’entraîner un codon stop prématuré dans le transcrit mature, celui-ci sera dégradé par le système de surveillance des ARN dans la cellule (nonsense mediated mRNA decay ou NMD) et la conséquence sera une absence de production de dystrophine dans le muscle des patients conduisant à un phénotype sévère de DMD. En revanche, si cette mutation permet de maintenir un cadre de lecture ouvert dans le transcrit muté, la synthèse d’une dystrophine tronquée en quantité normale ou réduite assurera un phénotype moins sévère de type BMD. Dans la majorité des cas de mutations ponctuelles et de délétions, cette règle s’applique, alors que la prédiction des conséquences des duplications est plus difficile.
Cette règle souffre cependant d’exceptions nombreuses mettant en exergue non seulement l’importance de la production d’une dystrophine « viable » mais aussi son caractère fonctionnel et la préservation de ses capacités à se lier avec ses principaux partenaires.
Les mutations prédites hors phase mais associées à un phénotype atténué
L’analyse des transcrits à partir de la biopsie musculaire permet d’expliquer comment des délétions mono-exoniques, comme par exemple celle de l’exon 45, prédites hors phase, ont été élargies en délétion en phase par un épissage alternatif de l’exon adjacent (ici l’exon 44) : la conséquence est la production minime d’une dystrophine suffisamment exprimée pour entraîner un phénotype plus modéré [7]. Dans la partie N-terminale du gène, la réactivation d’un promoteur alternatif situé dans l’exon 8 pourrait permettre de rendre « silencieuses » certaines des délétions situés en amont (ex : délétion des exons 3 à 7), permettant la synthèse d’une protéine tronquée mais fonctionnelle. Si un épissage alternatif affecte un exon porteur d’une mutation « non-sens » dans le domaine répété de la dystrophine (exons 23 à 42), une délétion en phase est alors générée et une dystrophine active produite. Une mutation non-sens très distale dans le gène (après l’exon 70) pourrait ne pas être reconnue par le système NMD, permettant la synthèse d’une protéine délétée de sa partie C-terminale mais toujours ancrée à la membrane par son site de liaison à la β-dystroglycane (exons 63-69).
Les mutations prédites en phase mais associées à un phénotype sévère
Lorsqu’une délétion exonique en phase touche un domaine fonctionnel essentiel comme celui du domaine de liaison à l’actine ou ABD1 au début du gène, le site d’ancrage de la protéine générée est alors aboli et le phénotype associé plus sévère. De la même façon, si l’étendue d’une délétion en phase débutant autour des exons 3 et 4 est trop large, on remarque souvent que le phénotype des patients qui en sont porteurs se rapproche davantage de celui d’une DMD que de celle d’une BMD. Un épissage alternatif aux bornes d’une délétion exonique en phase peut expliquer un phénotype sévère. La zone de liaison d’un des partenaires de la dystrophine, la nNOS, est codée par la partie du gène DMD située entre les exons 42 et 45. Elle se trouve abolie chez les patients porteurs d’une délétion en phase emportant les exons 45 à 55, théoriquement tous exprimant un phénotype BMD. L’étude d’une grande série de tels patients montre une hétérogénéité clinique corrélée à l’expression et à la localisation perturbée de cette nNOS [11] sans que l’on connaisse encore les mécanismes expliquant ces différences.
2. Les autres facteurs modificateurs
Certaines exceptions ne sont pas explicables en tenant compte uniquement des différents éléments cités plus haut reflétant ainsi la nécessité de mieux disséquer les mécanismes moléculaires liant le génotype au phénotype. Cela permettra peut-être d’aboutir à une règle plus universelle que la règle de Monaco. Durant ces dernières années, d’autres facteurs ont été identifiés comme pouvant influer sur la sévérité du phénotype au sein des dystrophinopathies.
-
Les gènes modificateurs : L’étude parallèle du transcriptome et du génome a permis d’impliquer plusieurs gènes dont des polymorphismes pourraient avoir un rôle dans la modulation du phénotype chez les patients souffrant deDMD[37]. Ainsi l’expression de l’ostéopontine, codée par le gène SPP1 dont le promoteur est activé par des protéines membres de la famille du TGF-β a pu être corrélée aux âges de perte de la marche variables chez les patients [28]. De même le rôle du TGF-β via la protéine LTBP4 (Latent TGF-β binding protein 4) est souligné dans des études de cohortes de patients dont la marche est perdue à des âges différents [10]. D’autres gènes modificateurs comme ANXA6 codant l’annexine A6, protéine liant le calcium et jouant un rôle dans la réparation musculaire a été impliqué chez les modèles animaux type Zebrafish mais pas encore chez l’homme. Chez les chiens GRMD (Golden Retriever muscular dystrophy), modèle canin de la DMD, comportant pourtant une absence complète de dystrophine, l’atteinte musculaire modérée et la longévité quasi normale peuvent être expliquées par une surexpression du gène Jagged1 codant une protéine régulatrice de la voie Notch [36].
-
La structure de la dystrophine : À l’état normal, le domaine central de la dystrophine comporte 24 répétitions, constituées d’environ 109 acides aminés chacune. Ces répétitions sont très peu identiques entre elles mais sont toutes caractérisées par la succession de motifs en heptades le long de la séquence. Une heptade est définie par sept acides aminés successifs numérotés de « a » à « g » où les résidus en position « a » et « d » sont hydrophobes. Les répétitions d’heptades sont la base de la structure en faisceau de trois hélices des répétitions de la dystrophine [16, 38]. Ainsi, chaque répétition est composée de trois hélices α reliées par deux boucles, le tout organisé en faisceau de trois hélices (coiled-coil) (Figure 2). Ces répétitions s’enchaînent et sont liées par une hélice commune formée par la troisième hélice de la première répétition et la première hélice de la suivante et dont la partie centrale est appelée linker (Figure 2).
 |
Figure 2. Structure en en faisceau de trois hélices de deux répétitions successives de la dystrophine. La répétition 17 comporte trois hélices αA, B et C) reliées par deux boucles. Elle s’enchaîne directement avec la répétition 18 (composée de trois hélices A’, B’ et C’) par son hélice C via un « linker ». |
La règle de Monaco a permis de comprendre la base des différences entre mutations qui décalent le cadre de lecture pour lesquelles il n’y a en général pas de dystrophine exprimée et un phénotype DMD associé, et celles conservant le cadre de lecture qui permettent l’expression de dystrophines tronquées, associées à un phénotype BMD réputé peu sévère. Le terme « partiellement fonctionnel » associé aux dystrophines tronquées est ainsi apparu comme le corollaire de l’adjectif « peu sévère » des phénotypes BMD. Cependant, même si cela reste une base importante, il convient de tempérer la règle de Monaco en examinant attentivement les cas de BMD avec diverses délétions exoniques. En effet, il y a des BMD sévères avec un âge de perte de marche et d’apparition d’une atteinte cardiaque relativement précoces. Nous avons fait l’hypothèse que les structures des différentes dystrophines tronquées ne sont peut-être pas similaires et pourraient en partie expliquer ces variations de sévérité. Nous avons ainsi récemment montré que la structure de la dystrophine résultant de délétions exoniques en phase dans le domaine central était corrélée à la sévérité des patients qui en sont porteurs (Figure 3) [26]. En effet, les délétions des exons 45 à 48 et 45 à 51, observées chez des patients BMD, entraînent la formation d’une nouvelle répétition (dite répétition hybride) ressemblant aux répétitions sauvages et sont responsables d’un phénotype moins sévère (en terme d’âge de perte de la marche et d’âge d’apparition d’une cardiomyopathie) que les délétions des exons 45 à 47 et 45 à 49 qui aboutissent à la formation d’une nouvelle structure ayant des caractéristiques éloignées de celle des répétitions sauvages (appelées répétition fractionnée) [26].
 |
Figure 3. Exemples des délétions des exons 45-47 et 45-55. La délétion 45-47 enlève la moitié de la répétition 17 et les 4/5es de la répétition 18 créant une nouvelle jonction (indiquée R17//R18) entre les séquences conservées de R17 et de R18. Cependant, cette séquence ne conserve pas la structure en coiled-coil de la région R17//R18. La délétion des exons 45-55 enlève la moitié de la répétition 17 et la moitié de la répétition R22 créant une nouvelle jonction entre les séquences conservées de ces deux répétitions. Cette séquence permet la reconstitution d’une structure en coiled-coil typique de la dystrophine. |
Rappel bref sur les banques de données dédiées aux dystrophinopathies et mise en place d’eDystrophin
Une banque de données est une collection de données organisées (génétiques et cliniques…) gérées par un système de gestion de banque de données (Excel, Access, UMD, LOVD…). Il existe actuellement des centaines de banques de données dédiées aux pathologies humaines et quelques dizaines consacrées aux affections neuromusculaires, ces dernières compilant en général les données génétiques et cliniques de patients, soit publiées dans la littérature, soit identifiées dans des laboratoires de diagnostic génétique de ces affections. Concernant les dystrophinopathies, la banque de données de Leiden (http://www.dmd.nl/nmdb/home.php?select_db=DMD_d) compile les mutations du gène DMD rapportées dans la littérature mais aussi celles soumises directement par les généticiens molécularistes qui le souhaitent. Depuis quelques années, de nombreux pays ont développé des banques de données dédiées à diverses affections neuromusculaires et plus particulièrement aux dystrophinopathies (voir liste exhaustive sur www.treat-nmd.eu/resources/patient-registries/list/). L’inclusion dans ces banques de données se fait soit par le professionnel de santé suivant le patient et/ou soit par un auto-enregistrement des patients via une interface internet. La banque de données UMD-DMD France (voir http://umd.be/DMD/), utilisant l’outil UMD comme système de gestion de banque de données, a été développée dans l’esprit de rassembler dans un lieu unique l’ensemble des données moléculaires et cliniques des patients dystrophinopathes analysés dans les 14 laboratoires de diagnostic des dystrophinopathies en France. L’accumulation de données concernant des milliers de patients permettrait non seulement d’établir des corrélations phénotype/génotype plus précises au sein des dystrophinopathies mais aussi de préparer l’arrivée des futurs traitements basés sur le gène. En effet, ces derniers s’appliquant sur des sous-groupes de patients porteurs de mutations spécifiques (délétions particulières, mutations non-sens), il était souhaitable que l’ensemble des patients français soient regroupés dans une banque de données unique.
Afin d’intégrer les conséquences structurales des mutations maintenant le cadre de lecture de la dystrophine, nous avons mis en place depuis 2012 une nouvelle banque de données appelée eDystrophin [25]. eDystrophin comporte un répertoire : (1) de toutes les mutations ne décalant pas la cadre de lecture, identifiées dans nos laboratoires ou rapportées dans la littérature, des phénotypes correspondants mais aussi des résultats de l’étude de la dystrophine en immunofluorescence et en Western-blot (WB) quand cette étude a été réalisée. Les délétions exoniques sont les plus fréquentes avec 61 % de toutes les mutations incluses dans eDystrophin, les duplications représentent 50 %, les mutations faux-sens 11 % et les petites délétions en phase 4 % et (2) des structures obtenues par modélisation par homologie des protéines résultant de ces délétions.
Cette banque de données pourrait permettre d’approfondir les corrélations phénotype/génotype en y associant un phénotype « structure protéique » apportant ainsi une meilleure connaissance des zones d’interaction de la dystrophine, plus proche de la réalité de la dystrophine dans le muscle. À terme, cela devrait donner des indications importantes pour les thérapies de la DMD par saut d’exon. Ce type de thérapies a pour but de transformer un patient DMD en patient BMD. Les conséquences structurales prédites dans eDystrophin pourraient permettre un meilleur choix des délétions cibles à obtenir avec le saut d’exon où il s’agit d’obtenir un phénotype BMD le moins sévère possible.
eDystrophin
1. Mise en place
eDystrophin, librement accessible via l’adresse http://edystrophin.genouest.org/, est une banque de données interactive développée avec MySQL 5.1.37 dans la suite logiciel MAMP (http://www.mamp.info/en/index.html). Le site web est construit avec XHTML, PHP 5 et JavaScript. La banque et le site sont hébergés par la plate-forme BioGenouest (http://www.genouest.org) et fonctionne avec le serveur Apache 2.2.3. My Domains (http://prosite.expasy.org/mydomains/) est utilisé pour représenter les protéines mutées et l’application Jmol (http://jmol.sourceforge.net/index.fr.html) est implémentée dans la banque pour la visualisation des structures tridimensionnelles des protéines.
En plus des données structurales concernant les différentes dystrophines issues de mutations non-tronquantes générées à l’Institut de Génétique et Développement de Rennes, eDystrophin contient des données cliniques et protéiques globales observées chez les patients porteurs de ces même mutations rapportés dans la littérature ou identifiés dans le laboratoire de biochimie et génétique moléculaire de l’hôpital Cochin. eDystrophin permet ainsi de confronter en un seul lieu les données structurales avec leurs conséquences cliniques et protéiques (Figure 4).
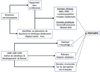 |
Figure 4. Sources du contenu actuel de la banque eDystrophin. |
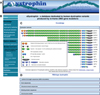 |
Figure 5. Accueil de la banque de données eDystrophin. (A) Bandeau d’accueil, (B) bandeau présentant les boutons interactifs permettant d’explorer la banque de données, (C) Présentation de la section « knowledge » montrant la sous-section « Dystrophin isoforms », (D) le bouton « Wild-type dystrophin » permet d’accéder à cinq sous-sections dans lesquelles le chercheur peut importer des données sur les isoformes de la dystrophine, les séquences des 79 exons, des domaines structuraux et des domaines d’interaction et enfin importer les structures ou les modèles tri-dimensionnels des différents domaines de la dystrophine. |
2. Rubriques
La banque eDystrophin est organisée en quatre sections : « Knowledge », « Explore database », « Statistics » et « Links » (Figure 5).
-
La rubrique « Knowledge » : Rassemble des connaissances de base sur le gène DMD, en donnant à l’aide d’un schéma les bornes des 79 exons, la position des 7 promoteurs internes et la position des différents domaines protéiques codés par les exons. Un schéma indique également les différents exons codant les différentes isoformes de la dystrophine. Deux sous-rubriques sont consacrées aux différentes isoformes protéiques en termes de domaine structuraux et aux différents domaines de liaison aux protéines et lipides partenaires.
-
La rubrique « Explore database » : Cette rubrique est essentiellement une rubrique de téléchargement de séquences ou d’informations concernant :
-
La dystrophine sauvage : isoformes, séquence exonique, domaines structuraux, domaines d’interactions, structure 3D de la dystrophine. Cette rubrique permet de télécharger toutes les données de base de séquences géniques ou protéiques, des domaines structuraux ou des domaines d’interaction avec les partenaires ainsi que les structures tridimensionnelles des domaines, obtenues soit par cristallographie soit par modélisation par homologie.
-
La dystrophine mutée (mutations en phase) ( Figures 6 et 7) Cette partie est le coeur et l’une des plus riches de la banque eDystrophin. Elle fait le répertoire de toutes les mutations en phase connues du gène DMD avec des indications sur le nombre de patients répertoriés et les origines des données (données issues du laboratoire de biochimie et génétique moléculaire [hôpital Cochin, Paris] ou données publiées), les résultats de l’étude de la dystrophine en WB ainsi que le modèle par homologie de toutes les délétions exoniques ne décalant pas le cadre de lecture.
Les requêtes se font soit par types mutationnels (délétions, duplications, mutations faux sens), soit par protéines résultant de tel ou tel saut d’exon, soit par phénotype (DMD, BMD, IMD, cardiomyopathie, asymptomatique ou inconnu) ou soit par domaine impliqué dans la mutation.

Figure 6. Exploration de la section « dystrophines mutées » de la banque de données. Après avoir activé le bouton délétions, le chercheur pointe sur ce schéma des exons du gène un exon impliqué dans la délétion d’intérêt. Apparaît le panneau à droite répertoriant les délétions impliquant cet exon. Le chercheur pointe alors la délétion d’intérêt. Activant le bouton « see protein » une sous-section s’ouvre (voir Figure 7).

Figure 7. Exemple de résultat de la requête pour la délétion des exons 45-47. La partie A renseigne sur la distribution phénotypique des patients porteurs de cette délétion inclus dans eDystrophin avec la liste de références bibliographiques des patients publiés. La liste de choix « Data displayed » permet d’afficher plusieurs paramètres phénotypiques ou les résultats de l’étude de la dystrophine en western blot. La partie B permet d’afficher le ou les domaines altérés par la mutation. La partie C permet d’afficher l’état altéré ou conservé des domaines d’interactions de la dystrophine. La partie D permet d’afficher entre autres une représentation de la structure tridimensionnelle obtenue par modélisation de la nouvelle jonction entre la répétition proximale et distale bordant la délétion. eDystrophin précise entre autres si une répétition « hybride » ou « fractionnée » est reconstituée.
-
-
La Rubrique « Statistics » : comporte quelques statistiques générales sur les données figurant dans eDystrophin.
-
La Rubrique « Links » : comporte des liens sur des sites utiles ainsi que la liste des références dont est extraite une partie des mutations incluses dans eDystrophin.
3. Utilisation
Nous présentons ci-dessous un exemple des données obtenues pour une délétion en phase très fréquente : la délétion des exons 45 à 47 (Figure 7).
Conclusion
Les dystrophinopathies représentent l’une des affections musculaires génétiques les plus fréquentes. La multiplicité de leurs expressions phénotypiques est sous-tendue non seulement par la diversité des mécanismes mutationnels mis en jeu dans le gène DMD mais aussi par les conséquences de ces mutations sur le transcrit et surtout sur la quantité et la qualité de la dystrophine finalement produite. Les modifications de la structure de la dystrophine résultant des mutations ne décalant pas le cadre de lecture interviennent dans la modulation fine du phénotype. La banque eDystrophin permet, par des requêtes simples, d’étudier les conséquences structurales des mutations DMD ne décalant pas le cadre de lecture. Nous encourageons la communauté médicale et scientifique à la consulter et l’utiliser pour mieux appréhender la variabilité des phénotypes observés mais aussi de mieux cibler les délétions d’intérêt dans le cadre de la thérapie basée sur le saut d’exon.
Liens d’intérêt
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Notez que l’acronyme « DMD » est utilisé pour désigner la maladie (DMD pour l’appellation anglo-saxonne « Duchenne Muscular Dystrophy ») dont les initiales se trouvent correspondre à l’appellation française « Dystrophie Musculaire de Duchenne ») et le gène DMD(qui comme tous les noms de gènes est conventionnellement écrit en italique).
Références
- Aartsma-Rus A, Van Deutekom JC, Fokkema IF, et al. Entries in the Leiden Duchenne muscular dystrophy mutation database: an overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve 2006 ; 34 : 135–144. [CrossRef] [PubMed] [Google Scholar]
- Amann KJ, Renley BA, Ervasti JM. A Cluster of basic repeats in the dystrophin rod domain binds F-actin through an electrostatic interaction. J Biol Chem 1998 ; 273 : 28419–28423. [CrossRef] [PubMed] [Google Scholar]
- Ayalon G, Davis JQ, Scotland PB, et al. An ankyrin-based mechanism for functional organization of dystrophin and dystroglycan. Cell 2008 ; 135 : 1189–1200. [CrossRef] [PubMed] [Google Scholar]
- Bhosle RC, Michele DE, Campbell KP, et al. Interactions of intermediate filament protein synemin with dystrophin and utrophin. Biochem Biophys Res Commun 2006 ; 346 : 768–777. [CrossRef] [PubMed] [Google Scholar]
- Campbell K, Kahl S. Association of dystrophin and an integral membrane glycoprotein. Nature 1989 ; 338 : 259–262. [CrossRef] [PubMed] [Google Scholar]
- Desguerre I, Laugel V. Diagnosis and natural history of Duchenne muscular dystrophy. Arch Pediatr 2015 ; 22 : 12S24–30. [CrossRef] [PubMed] [Google Scholar]
- Dwianingsih EK, Malueka RG, Nishida A, et al. A novel splicing silencer generated by DMD exon 45 deletion junction could explain upstream exon 44 skipping that modifies dystrophinopathy. J Hum Genet 2014 ; 59 : 423–429. [CrossRef] [PubMed] [Google Scholar]
- Emery AE. (ed). Diagnostic criteria for neuromuscular disorders baarn. The Netherlands : ENMC, 1994. [Google Scholar]
- Ervasti J, Campbell K. Membrane organization of the dystrophin-glycoprotein complex. Cell 1991 ; 66 : 1121–1131. [CrossRef] [PubMed] [Google Scholar]
- Flanigan KM, Ceco E, Lamar KM, et al. LTBP4 genotype predicts age of ambulatory loss in Duchenne muscular dystrophy. Ann Neurol 2013 ; 73 : 481–488. [CrossRef] [PubMed] [Google Scholar]
- Gentil C, Leturcq F, Ben Yaou R, et al. Variable phenotype of del45–55 Becker patients correlated with nNOSmu mislocalization and RYR1 hypernitrosylation. Hum Mol Genet 2012 ; 21 : 3449–3460. [CrossRef] [PubMed] [Google Scholar]
- Harper SQ, Hauser MA, DelloRusso C, et al. Modular flexibility of dystrophin: implications for gene therapy of Duchenne muscular dystrophy. Nat Med 2002 ; 8 : 253–261. [CrossRef] [PubMed] [Google Scholar]
- Koenig M, Hoffman EP, Bertelson CJ, et al. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell 1987 ; 50 : 509–517. [CrossRef] [PubMed] [Google Scholar]
- Koenig M, Monaco AP, Kunkel LM. The complete sequence of dystrophin predicts a rod-shaped cytoskeletal protein. Cell 1988 ; 53 : 219–226. [CrossRef] [PubMed] [Google Scholar]
- Lai Y, Zhao J, Yue Y, et al. Alpha2 and alpha3 helices of dystrophin R16 and R17 frame a microdomain in the alpha1 helix of dystrophin R17 for neuronal NOS binding. Proc Natl Acad Sci USA 2013 ; 110 : 525–530. [CrossRef] [Google Scholar]
- Le Rumeur E, Winder SJ, Hubert JF. Dystrophin: more than just the sum of its parts. Biochim Biophys Acta 2010 ; 1804 : 1713–1722. [CrossRef] [PubMed] [Google Scholar]
- Legardinier S, Hubert JF, Le Bihan O, et al. Sub-domains of the dystrophin rod domain display contrasting lipid-binding and stability properties. Biochim Biophys Acta 2008 ; 1784 : 672–682. [CrossRef] [PubMed] [Google Scholar]
- Legardinier S, Raguénès-Nicol C, Tascon C, et al. Mapping of the lipid-binding and stability properties of the central rod domain of human dystrophin. J Mol Biol 2009 ; 389 : 546–558. [CrossRef] [PubMed] [Google Scholar]
- Leturcq F, Tuffery-Giraud S. Genetics and molecular aspects of dystrophinopathies. Arch Pediatr 2015 ; 22 : 12S3–1. [CrossRef] [PubMed] [Google Scholar]
- Molza AE, Mangat K, Le Rumeur E, et al. Structural basis of neuronal nitric-oxide synthase interaction with dystrophin repeats 16 and 17. J Biol Chem 2015 ; 290 : 29531–29541. [CrossRef] [PubMed] [Google Scholar]
- Monaco A, Bertelson C, Liechti-Gallati S, et al. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics 1988 ; 2 : 90–95. [CrossRef] [PubMed] [Google Scholar]
- Monaco AP, Neve RL, Colletti-Feener C, et al. Isolation of candidate cDNAs for portions of the Duchenne muscular dystrophy gene. Nature 1986 ; 323 : 646–650. [CrossRef] [PubMed] [Google Scholar]
- Muntoni F, Torelli S, Ferlini A. Dystrophin and mutations: one gene, several proteins, multiple phenotypes. Lancet Neurol 2003 ; 2 : 731–740. [CrossRef] [PubMed] [Google Scholar]
- Newey SE, Benson MA, Ponting CP, et al. Alternative splicing of dystrobrevin regulates the stoichiometry of syntrophin binding to the dystrophin protein complex. Curr Biol 2000 ; 10 : 1295–1298. [CrossRef] [PubMed] [Google Scholar]
- Nicolas A, Lucchetti-Miganeh C, Ben Yaou R, et al. Assessment of the structural, functional impact of in-frame mutations of the DMD gene, using the tools included in the eDystrophin online database. Orphanet J Rare Dis 2012 ; 7 : 45. [CrossRef] [PubMed] [Google Scholar]
- Nicolas A, Raguenes-Nicol C, Ben Yaou R, et al. Becker muscular dystrophy severity is linked to the structure of dystrophin. Hum Mol Genet 2015 ; 24 : 1267–1279. [CrossRef] [PubMed] [Google Scholar]
- Petrof BJ, Shrager JB, Stedman HH, et al. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc Natl Acad Sci USA 1993 ; 90 : 3710–3714. [CrossRef] [Google Scholar]
- Piva L, Gavassini BF, Bello L, et al. TGFBR2 but not SPP1 genotype modulates osteopontin expression in Duchenne muscular dystrophy muscle. J Pathol 2012 ; 228 : 251–259. [CrossRef] [PubMed] [Google Scholar]
- Prins KW, Humston JL, Mehta A, et al. Dystrophin is a microtubule-associated protein. J Cell Biol 2009 ; 186 : 363–369. [CrossRef] [PubMed] [Google Scholar]
- Reynolds JG, McCalmon SA, Donaghey JA, et al. Deregulated protein kinase A signaling and myospryn expression in muscular dystrophy. J Biol Chem 2008 ; 283 : 8070–8074. [CrossRef] [PubMed] [Google Scholar]
- Rezniczek GA, Konieczny P, Nikolic B, et al. Plectin 1f scaffolding at the sarcolemma of dystrophic (mdx) muscle fibers through multiple interactions with beta-dystroglycan. J Cell Biol 2007 ; 176 : 965–977. [CrossRef] [PubMed] [Google Scholar]
- Sadoulet-Puccio H, Rajala M, Kunkel L. Dystrobrevin and dystrophin : an interaction through coiled-coil motifs. Proc Natl Acad Sci USA 1997 ; 94 : 12413–12418. [CrossRef] [Google Scholar]
- Sadoulet-Puccio HM, Khurana TS, Cohen JB, et al. Cloning and characterization of the human homologue of a dystrophin related phosphoprotein found at the Torpedo electric organ postsynaptic membrane. Hum Mol Genet 1996 ; 5 : 489–496. [CrossRef] [PubMed] [Google Scholar]
- Tuffery-Giraud S, Beroud C, Leturcq F, et al. Genotype-phenotype analysis in 2, 405 patients with a dystrophinopathy using the UMD-DMD database : a model of nationwide knowledgebase. Hum Mutat 2009 ; 30 : 934–945. [CrossRef] [PubMed] [Google Scholar]
- Van Deutekom JC, Bremmer-Bout M, Janson AA, et al. Antisense- induced exon skipping restores dystrophin expression in DMD patient derived muscle cells. Hum Mol Genet 2001 ; 10 : 1547–1554. [CrossRef] [PubMed] [Google Scholar]
- Vieira NM, Elvers I, Alexander MS, et al. Jagged 1 rescues the Duchenne muscular dystrophy phenotype. Cell 2015 ; 163 : 1204–1213. [CrossRef] [PubMed] [Google Scholar]
- Vo AH, McNally EM. Modifier genes and their effect on Duchenne muscular dystrophy. Curr Opin Neurol 28 : 528–534. [Google Scholar]
- Winder SJ, Gibson TJ, Kendrick-Jones J. Dystrophin and utrophin: the missing links! FEBS Lett 1995 ; 369 : 27–33. [CrossRef] [PubMed] [Google Scholar]
- Yamashita K, Suzuki A, Satoh Y, et al. The 8th and 9th tandem spectrin-like repeats of utrophin cooperatively form a functional unit to interact with polarity-regulating kinase PAR-1b. Biochem Biophys Res Commun 2010 ; 391 : 812–817. [CrossRef] [PubMed] [Google Scholar]
© R. Ben Yaou et al., publié par EDP Sciences, 2016
 Cet article est distribué sous licence « Creative Commons » : http://creativecommons.org/licenses/by/4.0/deed.fr/, permettant une ré-utilisation du contenu sans restriction à condition de mentionner clairement la source.
Cet article est distribué sous licence « Creative Commons » : http://creativecommons.org/licenses/by/4.0/deed.fr/, permettant une ré-utilisation du contenu sans restriction à condition de mentionner clairement la source.
Liste des figures
|
Figure 1. Les différents grands domaines de la dystrophine et les protéines interagissant avec elle. Voir texte pour les abréviations. |
| Dans le texte | |
|
Figure 2. Structure en en faisceau de trois hélices de deux répétitions successives de la dystrophine. La répétition 17 comporte trois hélices αA, B et C) reliées par deux boucles. Elle s’enchaîne directement avec la répétition 18 (composée de trois hélices A’, B’ et C’) par son hélice C via un « linker ». |
| Dans le texte | |
|
Figure 3. Exemples des délétions des exons 45-47 et 45-55. La délétion 45-47 enlève la moitié de la répétition 17 et les 4/5es de la répétition 18 créant une nouvelle jonction (indiquée R17//R18) entre les séquences conservées de R17 et de R18. Cependant, cette séquence ne conserve pas la structure en coiled-coil de la région R17//R18. La délétion des exons 45-55 enlève la moitié de la répétition 17 et la moitié de la répétition R22 créant une nouvelle jonction entre les séquences conservées de ces deux répétitions. Cette séquence permet la reconstitution d’une structure en coiled-coil typique de la dystrophine. |
| Dans le texte | |
|
Figure 4. Sources du contenu actuel de la banque eDystrophin. |
| Dans le texte | |
|
Figure 5. Accueil de la banque de données eDystrophin. (A) Bandeau d’accueil, (B) bandeau présentant les boutons interactifs permettant d’explorer la banque de données, (C) Présentation de la section « knowledge » montrant la sous-section « Dystrophin isoforms », (D) le bouton « Wild-type dystrophin » permet d’accéder à cinq sous-sections dans lesquelles le chercheur peut importer des données sur les isoformes de la dystrophine, les séquences des 79 exons, des domaines structuraux et des domaines d’interaction et enfin importer les structures ou les modèles tri-dimensionnels des différents domaines de la dystrophine. |
| Dans le texte | |
|
Figure 6. Exploration de la section « dystrophines mutées » de la banque de données. Après avoir activé le bouton délétions, le chercheur pointe sur ce schéma des exons du gène un exon impliqué dans la délétion d’intérêt. Apparaît le panneau à droite répertoriant les délétions impliquant cet exon. Le chercheur pointe alors la délétion d’intérêt. Activant le bouton « see protein » une sous-section s’ouvre (voir Figure 7). |
| Dans le texte | |
|
Figure 7. Exemple de résultat de la requête pour la délétion des exons 45-47. La partie A renseigne sur la distribution phénotypique des patients porteurs de cette délétion inclus dans eDystrophin avec la liste de références bibliographiques des patients publiés. La liste de choix « Data displayed » permet d’afficher plusieurs paramètres phénotypiques ou les résultats de l’étude de la dystrophine en western blot. La partie B permet d’afficher le ou les domaines altérés par la mutation. La partie C permet d’afficher l’état altéré ou conservé des domaines d’interactions de la dystrophine. La partie D permet d’afficher entre autres une représentation de la structure tridimensionnelle obtenue par modélisation de la nouvelle jonction entre la répétition proximale et distale bordant la délétion. eDystrophin précise entre autres si une répétition « hybride » ou « fractionnée » est reconstituée. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.