| Issue |
Cah. Myol.
Number 21, Juin 2020
|
|
|---|---|---|
| Page(s) | 36 - 41 | |
| Section | Myologie dans le monde / Myology around theworld | |
| DOI | https://doi.org/10.1051/myolog/202021013 | |
| Published online | 10 juillet 2020 | |
Chroniques russes...
Russian chronicles...
Institut de Myologie, Paris, France
* Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
L’école Euro-Russe de myologie a fêté l’an dernier son dixième anniversaire. Cette formation ras semble chaque année 100 à 150 spécialistes (neurologues, pédiatres, généticiens, rééducateurs et représentants de patients) venus des quatre coins de la Fédération de Russie et des pays de l’ancienne Union Soviétique (Figure 1). La myologie suscite, notamment parmi les jeunes générations de cliniciens, un engouement grandissant, surtout depuis que les laboratoires de biologie moléculaire à visée diagnostique se sont mis à utiliser les nouvelles techniques de séquençage à haut débit (NGS). L’errance diagnostique était jusqu’à il y a peu le lot de beau coup de malades neuromusculaires russes, faute de spécialistes suffisamment formés et informés, et du fait d’un réseau de soins globalement difficile d’accès, sauf dans les grandes métropoles comme Saint-Pétersbourg et Moscou.
 |
Figure 1 Les intervenants à la première Myo-Skola en 2010 à Saint Pétersbourg, avec autour de l’auteur, Ros Quinlivan, Jean-Thomas Vilquin, DmitryRudenko et Tima Stutcheskaya (agenouillés), Valery Kazakov et ses collègues. |
La Myo-Skola, puisque c’est le nom qu’on lui donne, se déroule généralement sur trois jours et alterne cours magistraux et ateliers pratiques. Elle a lieu le plus souvent à Saint-Pétersbourg, plus rarement à Moscou, et les frais d’inscription sont suffisamment modiques pour permettre la participation du plus grand nombre, y compris de ceux qui exercent dans des contrées reculées comme la Sibérie orientale ou la presqu’île du Kamtchatka (comptez sept heures d’avion entre Moscou et ces terres très lointaines...). Le corps enseignant est composé de spécialistes russes mais aussi de cinq à six invités européens (français, finlandais et allemands, pour l’essentiel), dont deux, membres fondateurs de la Myo-Skola, sont issus de l’Institut de Myologie de Paris. Le tout bénéficie d’un soutien fort utile et apprécié des associations de patients (Gaoordi et SMA-Russie, l’AFM-Téléthon) et de l’industrie pharmaceutique (Figures 2 et 3).
 |
Figure 2 La Myo-Skola 2014où l’auteur est entouré, entre autres, de Pierre Carlier et de Bjarne Udd. |
 |
Figure 3 La Myo-Skola 2019à Saint-Pétersbourg. La famille s’est agrandie... |
Les sessions interactives de cas cliniques constituent un temps fort de la Myo-Skola. À la différence de nos habitudes à l’Ouest, notamment en France, le patient et sa famille sont présents lors de la session et se prêtent d’autant plus au jeu qu’ils sont le plus souvent honorés d’avoir affaire, sans bourse délier, à des sommités du domaine. Certains viennent par fois de très loin pour bénéficier d’un deuxième avis, qu’il soit de nature diagnostique ou thérapeutique.
L’examen clinique se déroule, sauf exception, en public et chacun des participants peut poser, tout comme le patient, toutes les questions qu’il souhaite. On notera, avec un certain amusement, que la jeune génération des médecins russes s’est peu à peu affranchie des règles de soumission à l’autorité en n’hésitant plus à contredire les hypothèses, pour ne pas dire les « oukazes », diagnostiques proférées lors des premières Myo-Skolas par la vieille garde apparatchik locale.
Il nous faut aussi faire amende honorable quant aux hypothèses diagnostiques que nous-mêmes, tout auréolés de nos réputations de grands spécialistes de pathologie neuromusculaire venus de l’Ouest, avions avancées pour certains cas difficiles. L’erreur et l’ignorance sont et resteront humaines pendant encore longtemps...
Nous en voulons pour preuve l’observation clinique suivante qui s’est avérée très riche d’enseignements.
Une myotonie qui n’en était pas vraiment une...
En 2010, à l’occasion de la première édition de Myo-Skola, Viktoria Minina, une jeune interne de l’équipe de neurologie de Saint-Pétersbourg dirigée par Dmitry Rudenko et Tima Stutcheskaya, soumet à la sagacité des participants le cas très étrange d’une adolescente âgée de 18 ans prénommée Anastasia (Figure 4). Cette jeune fille née de parents bien portants a présenté, après un développement psychomoteur initial décrit comme normal, des troubles moteurs depuis l’âge de 7 ans. Ceci se traduit à l’époque par des difficultés à marcher ou courir sur la pointe des pieds, avec une faiblesse ressentie au niveau des jambes et des mains, ces dernières étant souvent l’objet de « crispations », notamment au froid. Plus occasionnellement, la patiente se plaint de myalgies relativement diffuses, sans caractère ni horaire particuliers. Rien enfin n’est notable dans l’histoire familiale : pas d’antécédents neuro musculaires en particulier, ni consanguinité. On signale juste des difficultés d’apprentissage à l’école qui l’ont empêchée d’accéder à l’enseignement supérieur. Elle a un frère cadet bien portant, et ellemême a donné naissance à un enfant bien portant il y a peu. Un bilan de débrouillage en neurologie aboutit à quelques éléments d’orientation : le taux sérique de CPK est légèrement augmenté à 1, 5 fois la normale, l’électro-neuromyogramme révèle la présence de décharges, qualifiées de pseudomyotoniques par l’électrophysiologiste, mais aussi d’un ralentissement des vitesses de conduction nerveuse motrices aux alentours de 38 à 40 m/s selon les segments de membres étudiés. On note une discrète atteinte sensitive. L’ensemble évoque plutôt un processus de dénervation chronique. L’imagerie cérébrale, les fonctions cardiaque et respiratoire sont rapportées comme normales. L’IRM des muscles des membres inférieurs et du bassin révèle des discrètes anomalies de signal au niveau du soléaire droit.
 |
Figure 4 Lors de la session interactive de la Myo-Skola 2010 : Viktoria Minina, Anastasia, la patiente, DmitryRudenko et le traducteur. |
Anastasia se prête volontiers à l’examen neurologique et au jeu des questions. L’examen met en évidence une crispation dynamique des doigts comparable à une myotonie. Ce point fera l’objet d’une discussion très animée entre les cliniciens présents. La marche est difficile, avec un discret steppage, et témoigne d’une atteinte plutôt distale. Les extenseurs des doigts sont également déficitaires. Les réflexes ostéo-tendineux sont abolis et le réflexe cutané plantaire est en flexion. Les paires crâniennes sont normales. Il n’y a pas de troubles sensitifs patents. Le visage est expressif, sans troubles de l’oculomotricité ni ptosis ni blépharospasme. La patiente est corpulente mais sans véritable hypertrophie musculaire à l’exception, peut-être, du mollet gauche (Figure 5). On ne note pas de pieds creux. La coexistence de cette myotonie ou pseudo myotonie, et d’éléments neuropathiques laisse l’assistance perplexe. Plusieurs hypothèses diagnostiques sont proposées mais aucune d’entre elles ne colle véritablement. Une dystrophie myotonique de Steinert parait peu probable tout comme une banale forme axonale de maladie de Charcot-Marie-Tooth. Une canalopathie est envisageable mais laquelle ? Une myopathie distale de type Laing, un moment évoquée, parait très improbable. Il est alors conseillé de refaire un EMG en appliquant le protocole élaboré par Emmanuel Fournier (CHU Pitié-Salpêtrière) pour l’exploration des canalopathies muscu laires, de faire un EMG aux deux parents, d’éliminer de principe une myotonie de Steinert en biologie moléculaire, et de continuer à suivre la patiente. On notera qu’à ce moment précis, les études génétiques étaient excessivement compliquées à organiser en Russie. D’abord parce que les autorités russes avaient décrété, depuis de nombreuses années, un embargo total sur l’envoi à l’étranger de tout test génétique à visée diagnostique. Ensuite parce que les laboratoires locaux, publics ou privés, n’étaient pas en mesure d’assurer ces prestations faute d’expertise et de matériel. Concernant les biopsies musculaires, la situation n’était guère plus enviable : très peu de centres experts ou prétendus tels, et une qualité technique très médiocre dans l’ensemble.
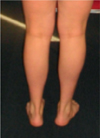 |
Figure 5 Les mollets cliniquement dans les limites de la normale. |
À l’époque, nous aurions sans doute pu avancer dans notre réflexion collégiale concernant cette énigme diagnostique si Bjarne Udd avait été là. Ce grand myologue finlandais de renommée internationale n’est en effet devenu un « invité permanent » et un des piliers de la Myo-Skola qu’à partir de l’année suivante (2011). Bjarne, qui combine les talents d’un clinicien hors pair et ceux d’un chercheur aguerri dans le domaine de la génétique moléculaire des maladies neuromusculaires, mais aussi de l’imagerie et de l’électrophysiologie, n’a pas son pareil, lors des sessions cliniques interactives, pour nous sortir des ornières découlant de l’errance diagnostique. Plusieurs myologues russes en herbe ont été formés à l’histologie musculaire par lui et son équipe du centre de référence neuromusculaire de Tampere. Quelques familles russes en quête d’un diagnostic plus précis ont également fait le déplacement, sou vent en voisins (n’oublions pas que la Finlande a longtemps subi la domination de la Russie).
Nous n’avons pas eu de nouvelles d’Anastasia jusqu’en mai 2019, date à laquelle nous fêtions les dix ans de la Myo-Skola à Saint-Pétersbourg. Elle faisait partie des patients à revoir lors d’une session interactive dédiée aux cold cases résolus. Son état clinique était resté stable tout comme sa gêne fonctionnelle. La carbamazépine, introduite de manière un peu empirique, avait eu une efficacité modérée sur la crispation des doigts. Entre temps, la patiente a pu bénéficier, dans un laboratoire moscovite privé, d’une étude en NGS réalisée sur un panel de gènes ciblant les neuropathies de type CMT et apparen tées. Cette étude a révélé la présence d’une mutation homozygote dans le gène HINT1 (c.110G>C; p.Arg37Pro ou R37P) ségrégeant parfaitement dans la famille. Le caractère pathogène de ce variant de séquence n’a pas été très difficile à établir, la mutation ayant déjà été rapportée en grand nombre à plusieurs endroits de l’immense territoire de la Fédération de Russie mais aussi en Europe de l’Est suggérant ainsi l’existence d’un effet fondateur dans les populations slaves.
À la lecture de la littérature sur les pathologies liées à HINT1, le tableau clinique était tout à fait compatible. Nous nous en voulons d’ailleurs de ne pas y avoir pensé plus tôt. À notre décharge, cette nou velle entité clinico-génétique, que l’on peut assimiler au groupe des neuromyotonies, n’a été formelle ment élucidée au niveau moléculaire qu’en 2012 [13]. Les premières descriptions les plus convaincantes de cette neuropathie rare remontent à Hahn en 1991 [4] même si d’autres auteurs avaient précédemment rapporté quelques observations similaires [3, 6].
D’un point de vue clinique, il ne s’agit pas d’une vraie myotonie mais d’une neuromyotonie. Les patients présentent certes une difficulté au relâche ment musculaire mais ont également une contraction musculaire persistante en dehors de l’effort, le tout sans l’aspect habituel EMG des canalopathies ni la pseudohypertrophie musculaire qui leur est souvent associée. Sur le plan électrophysio logique, la neuromyotonie correspond à une hyperexcitabilité du nerf périphérique. D’autres auteurs lui préfèrent le terme de décharges myokimiques. De façon générale, une neuromyotonie n’est pas toujours d’origine génétique, des étiologies auto immunes ayant été rapportées dans la littérature. L’autre élément-clé pour évoquer un diagnostic de neuromyotonie liée à HINT1 est la présence d’une neuropathie axonale de sévérité variable à nette pré dominance motrice ou motrice pure. Cette dernière peut d’ailleurs être isolée et donc révélatrice de l’affection.
Le gène HINT1 est responsable de trois principaux phénotypes au moins [1, 8, 10-12] : une forme axonale de CMT, une neuropathie distale motrice pure et une neuromyotonie comme dans l’observation rapportée ici. L’acronyme NMAN (pour neuromyo tonia and axonal neuropathy, MIM #137200) a été retenu par OMIM pour les désigner. D’un point de vue génétique, il existe au moins trois mutations fondatrices dans le monde (R37P, C84R, H112N) la première étant rapportée avec une grande fréquence dans les populations d’origine slave (Russie, République Tchèque, Slovénie) [2, 5, 7, 9]. L’évolution est très lentement progressive mais il existe des phénotypes plus sévères même avec un génotype identique. Un traitement par des molécules habituelle ment utilisées dans les neuromyotonies acquises comme la carbamazépine ou la phénylhydantoïne peut s’avérer efficace sur le phénomène pseudomyotonique mais peu ou pas sur le déficit moteur.
Le gène HINT1 code une enzyme, une phosphodimérase pour être précis, intervenant, sous la forme d’un homodimère, dans le métabolisme des purines. Ses substrats, et par voie de conséquence, ses fonctions, sont encore mal connus. Chez l’animal, la protéine semble surexprimée au niveau du nerf sciatique. HINT1 pourrait par ailleurs coder une protéine aux propriétés anti-tumorales.
Morale de l’histoire : la pathologie neuromusculaire s’enrichit chaque année de nouveaux phénotypes et de nouvelles anomalies génétiques. Lutter contre l’errance diagnostique fait partie de nos missions prioritaires de myologues et nous avons désormais des outils performants pour y parvenir. Dans un pays habitué, comme la Russie, aux grandes révolutions, l’avènement du NGS en constitue vraiment une dans le domaine de la génétique médicale. Les malades russes peuvent désormais directement en bénéficier même si son coût représente, pour beaucoup d’entre eux, une charge financière non négligeable. Myo-Skola, dont la prochaine édition aura lieu, si tout va bien, en octobre 2020 à Saint-Pétersbourg, a large ment contribué à informer les cliniciens russes de l’existence cette forme originale de neuromyotonie dans le monde slave. La mutation fondatrice R37P s’est avérée, et c’est d’ailleurs logique, être une cause majeure de CMT2 axonal en Russie [9] (Figure 6). Les Russes ne sont d’ailleurs pas peu fiers de cette trouvaille, et c’est tant mieux !
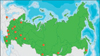 |
Figure 6 La répartition sur le territoire russe des cas de CMT2 axonal porteurs la mutation fondatrice slave du gène HINT1 (d’après Shchagina et al., 2020). |
Liens d’intérêt
L’auteur déclare n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Références
- Boaretto F, Cacciavillani M, Mostacciuolo ML, et al. Novel lossoffunction pathogenic variant of the HINT1 gene in a patient with distal motor axonal neuropathy without neuromyotonia. Muscle Nerve 2015 ; 52 : 688-9. [Google Scholar]
- Dadali EL, Nikitin SS, Kurbatov SA, et al. Clinical and genetic characteristics of autosomal recessive axonal neuropathy with neuromyotonia in Russian patients. Neuromuscul Dis 2017 ; 7 : 47-55. [CrossRef] [Google Scholar]
- Gamstorp I, Wohlfart G. A syndrome characterized by myokymia, myotonia, muscular wasting and increased perspiration. Acta Psychiat Neurol Scand 1959 ; 34 : 181-94. [CrossRef] [Google Scholar]
- Hahn AF, Parkes AW, Bolton CF, Stewart SA. Neuromyotonia in hereditary motor neuropathy. J Neurol Neurosurg Psychiatry 1991 ; 54 : 230-5. [CrossRef] [PubMed] [Google Scholar]
- Lasˇsˇuthová P, Brozˇková DSˇ, Kru°tová M, et al. Pathogenic variants in HINT1 are one of the most frequent causes of hereditary neuropathy among Czech patients and neuromyotonia is rather an underdiagnosed symptom. Neurogenetics 2015 ; 16 : 43-54. [Google Scholar]
- Mertens HG, Zschocke S. Neuromyotonie. Klin Wschr 1965 ; 43 : 917-25. [CrossRef] [Google Scholar]
- Peeters K, Chamova T, Tournev I, Jordanova A. Axonal neuropathy with neuromyotonia: there is a HINT. Brain 2017 ; 140 : 868-77. [PubMed] [Google Scholar]
- Rauchenzauner M, Frühwirth M, Hecht M, Kofler M, Witsch-Baumgartner M, Fauth C. A novel variant in the HINT1 gene in a girl with autosomal recessive axonal neuropathy with neuromyotonia: thorough neurological examination gives the clue. Neuropediatrics 2016 ; 47 : 119-22. [PubMed] [Google Scholar]
- Shchagina OA, Milovidova TB, Murtazina AF, et al. HINT1 gene pathogenic variants: the most common cause of recessive hereditary motor and sensory neuropathies in Russian patients. Mol Biol Rep 2020 ; 47 : 1331-7. [PubMed] [Google Scholar]
- Veltsista D, Chroni E. A first case report of HINT1-related axonal neuropathy with neuromyotonia in a Greek family. Clin Neurol Neurosurg 2016 : 148 : 85-7. [CrossRef] [PubMed] [Google Scholar]
- Wang Z, Lin J, Qiao K, et al. Novel pathogenic variants in HINT1 gene cause the autosomal recessive axonal neuropathy with neuromyotonia. Eur J Med Genet 2018 ; 62 : 190-4. [Google Scholar]
- Zhao H, Race V, Matthijs G, et al. Exome sequencing reveals HINT1 pathogenic variants as a cause of distal hereditary motor neuropathy. Eur J Hum Genet 2014 ; 22 : 847-50. [Google Scholar]
- Zimon´ M, Baets J, Almeida-Souza L, et al. Loss-of-function pathogenic variants in HINT1 cause axonal neuropathy with neuromyotonia. Nat Genet 2012 ; 44 : 1080-3. [Google Scholar]
© J.A. Urtizberea, publié par EDP Sciences, 2020
 Cet article est distribué sous licence « Creative Commons » : https://creativecommons.org/licenses/by/4.0/deed.fr, permettant une ré-utilisation du contenu sans restriction à condition de mentionner clairement la source.
Cet article est distribué sous licence « Creative Commons » : https://creativecommons.org/licenses/by/4.0/deed.fr, permettant une ré-utilisation du contenu sans restriction à condition de mentionner clairement la source.
Liste des figures
|
Figure 1 Les intervenants à la première Myo-Skola en 2010 à Saint Pétersbourg, avec autour de l’auteur, Ros Quinlivan, Jean-Thomas Vilquin, DmitryRudenko et Tima Stutcheskaya (agenouillés), Valery Kazakov et ses collègues. |
| Dans le texte | |
|
Figure 2 La Myo-Skola 2014où l’auteur est entouré, entre autres, de Pierre Carlier et de Bjarne Udd. |
| Dans le texte | |
|
Figure 3 La Myo-Skola 2019à Saint-Pétersbourg. La famille s’est agrandie... |
| Dans le texte | |
|
Figure 4 Lors de la session interactive de la Myo-Skola 2010 : Viktoria Minina, Anastasia, la patiente, DmitryRudenko et le traducteur. |
| Dans le texte | |
|
Figure 5 Les mollets cliniquement dans les limites de la normale. |
| Dans le texte | |
|
Figure 6 La répartition sur le territoire russe des cas de CMT2 axonal porteurs la mutation fondatrice slave du gène HINT1 (d’après Shchagina et al., 2020). |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.