| Issue |
Cah. Myol.
Number 15, Juin 2017
|
|
|---|---|---|
| Page(s) | 6 - 9 | |
| Section | Cas cliniques / Case reports | |
| DOI | https://doi.org/10.1051/myolog/201715002 | |
| Published online | 23 juin 2017 | |
Le Western-Blot à la rescousse…
The Western-Blot to the rescue…
1
CHU de Saint-Pierre, La Réunion, France
2
CHU Cochin, Paris, France
3
Centre de Référence, Hôpital Marin de Hendaye, Filnemus, Hendaye, France
Le diagnostic étiologique des dystrophies musculaires reste encore problématique, surtout à l’âge adulte. La situation se complique dès lors qu’on est à distance des premiers symptômes et que l’accès à des explorations paracliniques sophistiquées est ou était difficile.
Une observation déjà discutée lors d’une séance du Groupe d’Études en Myologie (GEM) de novembre 2015 (à l’occasion des JSFM de Lyon), est ici rapportée et commentée dans le détail.
Elle concerne deux cousins d’origine mahoraise suivis à la consultation du Centre de Référence neuromusculaire de l’Île de la Réunion (CHU de Saint-Pierre). Originaires de Mayotte, la partie française de l’archipel des Comores récemment devenue département d’outre-mer (DOM), ces deux patients présentent une histoire clinique superposable (Figures 1 et 2).
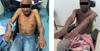 |
Figures 1 et 2 Les deux cousins atteints d’origine mahoraise présentant un déficit musculaire proximale lentement progressif. |
Le propositus a 40 ans. Il a deux frères restés à Mayotte et dont il n’a pas beaucoup de nouvelles. Il y aurait eu dans sa fratrie des enfants décédés en bas âge. Seul un cousin issu de la lignée maternelle, lui aussi installé à la Réunion (dans la région de Saint-Louis) présente des symptômes similaires (cf. infra). Malgré la forte endogamie régnant aux Comores, aucun lien formel de consanguinité n’a pu être établi au sein de cette famille.
Le patient rapporte des troubles moteurs depuis l’adolescence avec des difficultés à la course, à la montée des escaliers, et de façon plus générale une diminution de ses performances en sport. S’en est suivi au fil des ans une amyotrophie significative des deux cuisses plus particulièrement au niveau du muscle quadriceps. L’évolution est décrite comme lentement progressive. Suffisamment progressive et invalidante toutefois pour que le propositus ait recours à un fauteuil roulant à partir de l’âge de 33 ans. Le déficit prédomine alors aux muscles des racines, surtout pelvienne et, dans une moindre mesure, scapulaire. La fonte musculaire est plus marquée au niveau des quadriceps, des muscles des avant-bras et des pectoraux. Elle contraste avec une discrète hypertrophie notée au niveau des mollets et à un degré moindre au niveau de la langue. L’atrophie partielle du biceps brachial fait évoquer une « boule » telle que décrite par Michel Fardeau dans les dysferlinopathies. Il n’y a pas ou peu de rétractions musculotendineuses (Figure 3).
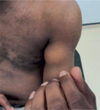 |
Figure 3 L’aspect en boule du biceps brachial a fait évoquer un moment une dysferlinopathie. |
À 38 ans, il est encore capable de se verticaliser et de marcher jusqu’à 500 mètres avec de l’aide. Les quelques examens complémentaires réalisés au début de la maladie ont simplement permis d’affirmer le caractère myopathique des troubles, sans plus de précision. Le taux sérique de la créatine-phospho-kinase (CPK) a été d’emblée élevé, autour de 2 000 unités par litre. En 2002, à l’âge de 26 ans, une biopsie musculaire est réalisée à St-Denis de la Réunion au niveau du muscle quadriceps gauche. Elle ne révèle que des lésions myopathiques et non spécifiques, tandis que l’immunocytochimie s’avère non concluante. Il existe par ailleurs une discrète diminution de la capacité vitale (74%), avec un syndrome obstructif associé, le tout étant mis sur le compte de séquelles anciennes d’une dilatation chronique des bronches. Le cardiologue note une légère hypokinésie du ventricule gauche associée à une discrète hypertrophie. Un traitement par IEC va permettre de la stabiliser complètement. L’IRM musculaire est réalisée à un âge assez avancé, à 38 ans, et montre une atteinte diffuse, peu sélective et se révèle non contributive. Seul le muscle couturier (sartorius) est relativement préservé (Figures 4 et 5).
 |
Figures 4 et 5 L’IRM musculaire des membres inférieurs montre des lésions dégénératives déjà très avancées. |
Le cousin, âgé de 48 ans, a perdu un frère décédé à l’âge de 16 ans de problèmes supposément d’origine cardiaque. Il a onze demi-frères et sœurs du côté du père qui sont en bonne santé. Il a eu trois enfants qui sont décrits comme bien portants. Les premiers symptômes (fatigue, difficultés à la marche et à monter les escaliers, tiraillements dans les mollets) sont apparus au début de l’adolescence. Le déficit est également proximal. Il a eu recours à un fauteuil roulant à partir de 45 ans mais est encore capable de faire quelques pas avec un déambulateur. L’amyotrophie est assez diffuse et les rétractions tendineuses peu développées. On ne note pas d’atrophie sélective du biceps brachial. Le galbe des mollets est plutôt fin (Figures 6 et 7). Il existe une atteinte des membres supérieurs (réduction de l’abduction à 60°) mais sans décollement manifeste des omoplates. Le taux de CPK est comparable à celui de son cousin, autour de 2 à 3 000 unités par litre. Le bilan cardiaque est rassurant, y compris en échographie, tout comme le bilan respiratoire. Le patient a eu deux biopsies musculaires dont la dernière en 2001. Un doute existait concernant l’intensité du marquage les anticorps DYS 2 et DYS3 dirigés contre la dystrophine. Un ré-examen des lames en métropole à l’aide de techniques immunologiques plus sensibles (de type DAB) a toutefois conclu à la normalité des immunomarquages contre les trois épitopes de la dystrophine, permettant ainsi d’écarter l’hypothèse d’une dystrophinopathie.
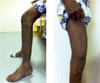 |
Figures 6 et 7 La gracilité relative de la musculature des mollets avait fait évoquer initialement une myopathie distale de type dysferlinopathie. |
Ces deux patients issus de la même famille ont été vus à plusieurs reprises lors de missions neuromusculaires venant de métropole. Le diagnostic de dysferlinopathie, suggéré par l’histoire, la clinique et la présence d’une atrophie bicipitale sélective, a longtemps été évoqué jusqu’au moment où l’analyse complète du gène DYSF et la normalité des immunomarquages contre cette dernière a permis d’éliminer formellement cette hypothèse. Les quatre gènes codant les différentes sarcoglycanes ont aussi été séquencés, également en vain. De même pour le gène LMNA du fait de la possible association avec un phénotype cardiaque.
Dans la situation d’impasse dans laquelle cette famille se trouvait, et en préalable à d’éventuelles études de séquençage de gènes à haut débit, une nouvelle biopsie musculaire a été proposée au propositus. Réalisée au niveau d’un deltoïde, celle-ci a montré des lésions assez peu spécifiques faites d’inégalité de calibre des fibres, d’internalisations nucléaires et de fibres musculaires, assez peu nombreuses, en nécrose-régénération. Les immunomarquages standards pour la dystrophine et les autres protéines membranaires se sont à nouveau avérés normaux. Seule était noté une discrète sur-expression de l’utrophine.
L’élucidation du diagnostic est finalement venue du Western-Blot. Réalisé à l’hôpital Cochin sur un fragment musculaire transféré depuis l’Ile de la Réunion, cette méthode basée sur la migration des protéines en fonction de leur poids moléculaire, a objectivé une réduction de la quantité de dystrophine mais surtout une augmentation de taille de la protéine rendant hautement probable le diagnostic de dystrophinopathie. À noter que les sarcoglycanes étudiés sur ce même Western-Blot dit multiplex étaient également légèrement diminués, un phénomène secondaire bien connu et souvent observé dans les dystrophinopathies primitives. Une analyse par PCR Multiplex semi-quantitative du gène DMD (MLPA) est venue confirmer cette hypothèse en mettant en évidence une duplication en phase, respectant donc le cadre de lecture, d’une région allant des exons 14 à 29. Les deux cousins sont bien porteurs de la même duplication et une analyse de ségrégation est cours dans le reste de la famille (Figures 8 et 9).
 |
Figures 8 et 9 Images du Western-Blot où l’on visualise parfaitement la diminution de taille du signal de la dystrophine. |
Discussion
Cette observation est riche d’enseignements à plus d’un titre. Elle démontre d’abord l’intérêt du Western-Blot dans l’évaluation des dystrophies musculaires, surtout dans la myopathie de Becker où l’mmunomarquage sur coupes congelés est parfois pris en défaut. Cette technique semi-quantitative n’est toutefois pas systématiquement demandée a fortiori si les lésions primitives paraissent bénignes ou peu importantes. Elle est difficilement envisageable en routine dans le contexte des Centres de Référence neuromusculaires ultramarins car elle nécessite, pour parvenir aux quelques laboratoires métropolitains qui la proposent, que le fragment de muscle voyage congelé.
Dans le cas présent, l’attention aurait dû être plus attirée par la cardiomyopathie présente chez l’un des deux cousins. Il est vrai que celle-ci est apparue plus tardivement dans l’évolution. Le lien de causalité avec le décès prématuré d’un des frères du cousin âgé de 16 ans reste et restera purement spéculatif. À notre connaissance, la cardiomyopathie dans la BMD est rarement aussi précoce et évolutive.
L’imagerie n’a pas été ici très contributive du fait du stade avancé des lésions mais dans d’autres circonstances, elle peut s’avérer très utile.
La duplication du gène DMD identifiée dans cette famille (duplication des exons 14-29) a déjà été rapportée dans la base de données française UMD-DMD (www.umd.be/DMD/). Trois patients au moins y sont répertoriés avec à chaque fois un phénotype BMD. L’un d’entre eux a bénéficié d’une transplantation cardiaque.
La myopathie de Becker est souvent source d’erreurs diagnostiques. Elle peut être confondue avec l’ensemble, très hétérogène cliniquement et génétiquement, des dystrophies musculaires dite des ceintures (LGMD pour Limb Girdle Muscular Dystrophy en anglais) La variabilité du phénotype Becker à l’intérieur d’une même famille, voire d’une même fratrie, reste également fréquente. Le fait que le cousin plus âgé soit, à 48 ans, encore indemne de toute atteinte cardiaque en est la preuve.
À l’heure actuelle, et faute d’avoir recours à une biopsie musculaire d’emblée, on aurait tendance, devant une histoire clinique de la sorte (déficit musculaire proximal, élévation franche des CPK, atteinte cardiaque même minime, généalogie compatible avec une transmission récessive liée au chromosome X), à prescrire d’emblée une étude gène DMD en MLPA suivie, en cas de négativité, d’une étude sur un panel de gènes associant myopathie et atteinte cardiaque. Dans les situations d’impasse diagnostique, le recours au Western-Blot s’avère bien utile et permet d’explorer plusieurs pistes étiologiques à la fois. Le Western-Blot reste néanmoins une méthode difficilement exportable, surtout dans des centres où l’activité de biopsie musculaire est faible [1, 2].
Il convient aussi de rappeler que même dans un environnement fortement endogamique, la dystrophie musculaire la plus fréquente reste la myopathie de Duchenne et, avec une fréquence moindre, à la myopathie de Becker.
La découverte du gène causal au sein de cette famille va enfin permettre de prodiguer un conseil génétique éclairé et fiable. Un dépistage des patients à risque au niveau cardiaque permettra de mieux prévenir les complications et le cas échéant de les traiter [3]. De manière générale, les transmettrices BMD peuvent en effet, de manière générale, être à risque de présenter une cardiomyopathie. Dans cette famille, et fort heureusement pour elles, les deux mères respectives des deux cousins sont décrites comme bien portantes.
Liens d’intérêt
L’auteur déclare n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Références
- Leturcq F, Tuffery-Giraud S. Genetics and molecular aspects of dystrophinopathies. Arch Pediatr 2015; 22 (suppl 1) : 12S3–11. [CrossRef] [PubMed] [Google Scholar]
- Soltanzadeh P, Friez MJ, Dunn D, et al. Clinical and genetic characterization of manifesting carriers of DMD mutations. Neuromuscul Disord 2010; 20 : 499–504. [PubMed] [Google Scholar]
- Aartsma-Rus A, Ginjaar IB, Bushby K. The importance of genetic diagnosis for Duchenne muscular dystrophy. J Med Genet 2016; 53: 145–51. [PubMed] [Google Scholar]
© A. Choumert et al., publié par EDP Sciences, 2017
 Cet article est distribué sous licence « Creative Commons » : http://creativecommons.org/licenses/by/4.0/deed.fr/, permettant une ré-utilisation du contenu sans restriction à condition de mentionner clairement la source.
Cet article est distribué sous licence « Creative Commons » : http://creativecommons.org/licenses/by/4.0/deed.fr/, permettant une ré-utilisation du contenu sans restriction à condition de mentionner clairement la source.
Liste des figures
|
Figures 1 et 2 Les deux cousins atteints d’origine mahoraise présentant un déficit musculaire proximale lentement progressif. |
| Dans le texte | |
|
Figure 3 L’aspect en boule du biceps brachial a fait évoquer un moment une dysferlinopathie. |
| Dans le texte | |
|
Figures 4 et 5 L’IRM musculaire des membres inférieurs montre des lésions dégénératives déjà très avancées. |
| Dans le texte | |
|
Figures 6 et 7 La gracilité relative de la musculature des mollets avait fait évoquer initialement une myopathie distale de type dysferlinopathie. |
| Dans le texte | |
|
Figures 8 et 9 Images du Western-Blot où l’on visualise parfaitement la diminution de taille du signal de la dystrophine. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.