| Issue |
Cah. Myol.
Number 25, juillet 2022
|
|
|---|---|---|
| Page(s) | 23 - 29 | |
| Section | Mise au point / Focus | |
| DOI | https://doi.org/10.1051/myolog/202225006 | |
| Published online | 11 août 2022 | |
Les myopathies oculo-pharyngo-distales : des nouvelles maladies à expansions de répétitions CGG
Oculopharyngeal distal myopathies: new diseases with CGG repeats expansions
1
Institut de Génétique et de Biologie Moléculaire et Cellulaire (IGBMC) Inserm U 1258, CNRS UMR 7104, Université de Strasbourg, 67404 Illkirch, France
2
Department of Neurology, Peking University First Hospital, Beijing 100034, China
* Contact Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Résumé
La myopathie oculo-pharyngo-distale (OPDM) est une maladie génétique rare de l’adulte affectant les muscles squelettiques du visage, du pharynx et des extrémités des membres. Récemment, des variants dans quatre gènes distincts ont été identifiés comme responsables de cette pathologie. Bien que localisées dans différents gènes, le mécanisme mutationnel est identique, à savoir une expansion de 50 à 200-300 répétitions de triplets de nucléotides CGG. Dans cet article, nous décrivons les aspects cliniques, histopathologiques et génétiques de l’OPDM, ainsi que les mécanismes moléculaires pouvant expliquer la toxicité de ces expansions de répétitions trinucléotidiques.
© M. Boivin et al., publié par EDP Sciences, 2022
 Cet article est distribué sous licence « Creative Commons » : http://creativecommons.org/licenses/by/4.0/deed.fr/, permettant une ré-utilisation du contenu sans restriction à condition de mentionner clairement la source.
Cet article est distribué sous licence « Creative Commons » : http://creativecommons.org/licenses/by/4.0/deed.fr/, permettant une ré-utilisation du contenu sans restriction à condition de mentionner clairement la source.
Caractéristiques cliniques et ThistopatThologiques de l’OPDM
La myopathie oculo-pharyngo-distale (OPDM, OMIM 164310) est une maladie génétique autosomique dominante rare touchant les muscles squelettiques et se manifestant à l’âge adulte. Depuis la première description de cette maladie en 1977, environ 300 patients atteints d’OPDM ont été recensés dans le monde, surtout en Asie (Chine, Japon, Corée du Sud), mais aussi en Europe et aux États-Unis [1, 2]. D’un point de vue clinique, l’OPDM est caractérisée par un ptosis progressif, une ophtalmoplégie, une dysphagie, une dysarthrie, ainsi qu’une faiblesse des muscles faciaux, pharyngés et des membres distaux. Cette atteinte musculaire est associée à une augmentation modérée du taux de créatine kinase sérique, une insuffisance respiratoire précoce d’intensité variable et un risque accru de problèmes cardiovasculaires sur le long terme. L’âge moyen d’apparition de la maladie est estimé à 28 ans, plus ou moins 11 ans, le phénotype complet de l’OPDM s’étalant généralement sur une dizaine d’années [3]. D’un point de vue histopathologique, on constate une fibrose endomysiale, une variation de la taille des fibres musculaires, des vacuoles cytoplasmiques et un remplacement, plus ou moins marqué, par du tissu fibro-adipeux (Figures 1A, 1B). Les fibres musculaires présentent aussi des spécificités propres à l’OPDM telles que la présence d’inclusions intranucléaires éosinophiliques de 1 à 2 μm de diamètre, positives pour les protéines p62 et ubiquitine (Figure 1C), donnant un aspect tubulo-filamentaire en microscopie électronique [4, 5].
 |
Figure 1 Caractéristiques histopathologiques de biopsies de patients atteints d’OPDM. (A, B) Présence de vacuoles cytoplasmiques révélées par une coloration (A) au trichrome de Gomori ou (B) à l’hématoxyline-éosine et (C) d’inclusions intranucléaires positives pour la protéine p62. |
Plusieurs anomalies génétiques responsables de l’OPDM
Les progrès du séquençage du génome humain, et tout particulièrement la technique, très originale, dite de « long read », permettant de lire de très longues séquences d’ADN, ont abouti à l’identification de plusieurs causes génétiques à l’origine de l’OPDM (Figure 2). En 2019, une expansion anormale de répétitions de triplets de nucléotides CGG a été identifiée dans la région 5’UTR du gène LRP12 chez 22 patients japonais [6]. Cette expansion dans le gène LRP12 n’a été retrouvée que chez approximativement 40% des cas d’OPDM seulement, suggérant l’existence d’une hétérogénéité génétique dans cette maladie. De fait, en 2020, Deng et ses collaborateurs ont montré qu’une expansion anormale de répétitions CGG dans le 5’UTR du gène GIPC1 était associée à l’OPDM chez 12 patients chinois [2]. Cette découverte a ensuite été confirmée par une seconde étude réalisée sur une cohorte encore plus grande de patients OPDM [7]. En 2021, c’est une expansion de répétitions CGG dans la région 5’UTR du gène NOTCH2NLC qui a été identifiée [8]. Enfin, en 2022, une expansion CGG dans la région 5’UTR du gène RILPL1 a été associée à l’OPDM [9].
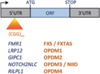 |
Figure 2 Les expansions de répétitions CGG impliquées dans des pathologies. Des expansions de répétitions CGG identifiées dans les régions 5’UTR des gènes FMR1, LRP12, GIPC1, NOTCH2NLC et RILPL1 sont responsables du syndrome de l’X fragile (FXS) ou du syndrome de tremblement et d’ataxie lié à l’X fragile (FXTAS), de la maladie à inclusions intranucléaires neuronales (NIID) et de la myopathie oculo-pharyngo-distale (OPDM). |
Quel que soit le gène hôte, un nombre de répétitions CGG inférieur à 30 a été détecté chez les sujets sains, tandis que les patients atteints d’OPDM présentaient une expansion de répétitions comprise entre 60 et 300 [7]. L’OPDM est ainsi divisée en 4 sous-types : l’OPDM1 (OMIM 164310) lié à LRP12, l’OPDM2 (OMIM 618940) lié à GIPC1, l’OPDM3 (OMIM 619473) lié à NOTCH2NLC et, l’OPDM4 (OMIM 619790) à RILPL1 (Figure 2). Ces quatre sous-types sont cliniquement et histologiquement superposables et partagent le même mécanisme mutationnel. Ces données suggèrent que les expansions de répétitions CGG jouent un rôle essentiel dans la pathogenèse de l’OPDM, et ceci indépendamment des gènes dans lesquels ces répétitions se trouvent. Il reste toutefois à éclaircir comment ces expansions de répétitions CGG conduisent au développement de cette pathologie.
Expansion de répétitions nucléotidiques et maladies neuromusculaires
À ce jour, il existe une cinquantaine de maladies neurodéveloppementales, neurodégénératives et/ ou neuromusculaires causées par des expansions de répétitions nucléotidiques. Parmi les maladies neuromusculaires, nous citerons les dystrophies myotoniques de type 1 et 2 (DM1, OMIM 160900 / DM2, OMIM 602668) et la dystrophie musculaire oculopharyngée (OPMD, OMIM 164300). Compte tenu de leur mécanisme génétique proche et d’une symptomatologie en partie superposable entre ces pathologies et l’OPDM, on peut se demander si ces maladies ne partagent pas les mêmes mécanismes moléculaires de toxicité.
Les dystrophies myotoniques de type 1 et 2 (DM1/DM2) sont caractérisées par une faiblesse et une atrophie musculaires, une myotonie, une atteinte cardiaque, une cataracte, des troubles cognitifs ainsi qu’une résistance à l’insuline. La DM1 est causée par une expansion de quelques centaines à plusieurs milliers de répétitions de tri-nucléotides CTG située dans la région 3’UTR du gène DMPK [11], tandis que la DM2 est due à une expansion de 75 à 11 0000 répétitions quadri-nucléotidiques CCTG dans le premier intron du gène CNBP [11]. Ces répétitions CTG/CCTG sont transcrites en ARN contenant alors un nombre anormalement élevé de répétitions CUG/CCUG lesquelles forment des foci d’ARN dans le noyau des cellules. D’un point de vue moléculaire, il a été montré que les protéines « Muscle blind-like 1/2 » (MBNL1 et MBNL2) se lient spécifiquement aux répétitions CUG et qu’elles colocalisent avec les foci d’ARN CUG/CCUG [12–14]. La séquestration des protéines MBNL1/2 dans les foci d’ARN entraîne une perte de fonction de ces facteurs d’épissage, conduisant à des altérations de l’épissage de nombreux autres ARN expliquant ainsi les multiples symptômes des patients atteints de DM. Un telmécanisme reste peu probable dans l’OPDM puisque les expansions de répétitions CGG dans les gènes LRP12, GIPC1, NOTCH2NLC et RILPL1 sont nettement plus courtes (de 60 à 200-300 répétitions) que chez les patients atteints de DM et qu’aucun agrégat d’ARN nucléaire n’a été observé dans les cellules des patients atteints d’OPDM.
-
La dystrophie musculaire oculo-pharyngée (OPMD) est une pathologie musculaire qui se rapproche le plus, d’un point de vue clinique et histologique, de l’OPDM, avec un ptosis, une atteinte musculaire proximale, une dysphagie et des difficultés respiratoires. Les caractéristiques histopathologiques sont similaires comme la présence de vacuoles bordées dans des fibres musculaires par ailleurs atrophiées, et la présence d’inclusions intranucléaires [15]. En revanche, le début de cette affection est très tardif, généralement au-delà de l’âge de 50 ans. L’OPMD est elle aussi causée par une expansion de répétitions de tri-nucléotides GCG, mais cette expansion est bien plus courte que dans le cas de l’OPDM avec une extension de 1 à 8 répétitions GCG situées dans la phase codante du gène PAPBN1 codant la protéine PABP2. Cette expansion de répétitions conduit à une protéine PABP2 avec un nombre étendu d’acides aminés alanines dans sa partie N-terminale (jusqu’à 18 alanines alors que dans la population générale on dénombre 10 alanines) [16]. Cette extension de résidus polyalanines conduit à une perte de fonction et/ou un gain de fonction dominant négatif sur l’activité de la protéine PABP2 [17, 18]. Toutefois, les expansions de répétitions CGG dans les gènes LRP12, GIPC1, NOTCH2NLC et RILPL1 étant plus longues (de 60 à 200-300 répétitions) et localisées dans les régions 5’UTR non traduites de ces gènes, un mécanisme de toxicité commun entre l’OPDM et l’OPMD semble également peu probable.
Les mécanismes moléculaires impliqués dans la toxicité de ces expansions de répétitions CGG dans l’OPDM sont donc, à ce jour, largement inconnus. La nature de la mutation n’est toutefois pas sans rappeler celle observée dans d’autres maladies, tel que le syndrome de tremblement et d’ataxie lié à l’X fragile (FXTAS) et la maladie à inclusions intranucléaires neuronales (NIID), dont les mécanismes toxiques sous-jacents ont été élucidés il y a peu. En effet, avant l’identification des expansions de répétitions CGG dans les OPDM de type 1 à 4, des mutations similaires en composition (CGG), en taille (60 à 200-300 répétitions) et en localisation (5’UTR) ont été impliquées dans d’autres pathologies. En effet, des expansions de 50 à 200-300 répétitions des tri-nucléotides CGG dans les régions 5’UTR des gènes FMR1 et NOTCH2NLC sont responsables respectivement du syndrome de tremblement et d’ataxie lié à l’X fragile (FXTAS, OMIM 300623) et de la maladie à inclusions intranucléaires neuronales (Figure 2; NIID, OMIM 603472).
-
Le syndrome de tremblement et d’ataxie lié à l’X fragile [1, 2] (FXTAS) est une maladie neurodégénérative caractérisée par une démarche ataxique et/ou un tremblement intentionnel; les patients peuvent également présenter des altérations neuropsychiatriques et cognitives ainsi que des symptômes parkinsoniens [19]. Cette maladie touche principalement les hommes et apparaît généralement entre 60 et 65 ans. D’un point de vue histopathologique, l’analyse des cerveaux de patients FXTAS montre une diminution du nombre de cellules de Purkinje, une gliose des cellules de Bergmann ainsi qu’une spongiose de la substance blanche des pédoncules moyens du cervelet [20, 21]. De plus, des inclusions intranucléaires éosinophiliques et positives à l’ubiquitine et p62, sont retrouvées dans les neurones et les astrocytes, mais aussi dans de nombreux autres tissus à l’exception des muscles squelettiques, où le gène FMR1 n’est pas exprimé [21, 22]. Le FXTAS est causé par une expansion de 55 à 200 répétitions de tri-nucléotides CGG dans la région 5’UTR du gène FMR1 (Figure 2) [23]. Cette mutation n’est pas sans rappeler la cause du syndrome de l’X fragile (FXS, OMIM 300624), où des expansions de plus de 200 répétitions CGG conduisent à une hyperméthylation du promoteur du gène FMR1, entraînant une perte d’expression de ce gène et donc une perte d’expression de la protéine FMRP codée par ce gène [24, 25]. Le déficit en protéine FMRP est responsable d’un retard mental et de troubles autistiques, qui sont caractéristiques du syndrome de l’X fragile. Néanmoins, contrairement à FXS, où ces expansions excèdent 200 répétitions de nucléotides CGG, FXTAS présente un nombre de répétitions plus faible, qu’on appelle alors prémutation. Cette prémutation ne conduit pas à une hyperméthylation du promoteur du gène FMR1, et les patients FXTAS présentent donc une expression normale de ce gène et de la protéine FMRP [26]. Ces données moléculaires permettent ainsi d’écarter l’hypothèse d’une perte d’expression du gène FMR1 dans FXTAS. Par la suite, il a été démontré que, malgré la localisation des expansions de répétitions CGG dans une région prédite originellement comme non-codante (5’UTR), ces expansions sont tout de même traduites en une protéine composée de polyglycines et appelée FMRpolyG [27, 28]. En effet, cette expansion de répétitions CGG est située dans un petit cadre de lecture ouvert (ORF pour « Open Reading Frame »), situé en amont de l’ORF principale codant FMRP, et où chaque triplet de CGG code pour un acide aminé glycine. Il est intéressant de noter que l’initiation de la traduction de cette petite ORF n’a pas lieu à un codon classique ATG, mais à un codon ACG [28]. En effet, l’initiation à des codons proches du codon ATG, tels que des codons CTG, GTG TTG ou ACG, permet une initiation faible de la traduction par mésappariement avec le codon initiateur de l’ARNt et sont alors décodés en méthionine [29]. Cette nouvelle protéine, FMRpolyG, forme des inclusions intranucléaires typiques dans les modèles cellulaires et animaux de la FXTAS, et son expression est suffisante pour induire un phénotype ataxique chez les souris [27, 28]. Enfin, le développement d’anticorps dirigés contre la protéine FMRpolyG et l’analyse de tissus de patients FXTAS a permis de confirmer la présence de cette protéine dans les inclusions intranucléaires typiques de cette maladie (Figure 3A).

Figure 3 Inclusions intranucléaires dans FXTAS et NIID. Immunofluorescence sur des lames de cervelet (A) d’un patient FXTAS montrant des inclusions intranucléaires de protéine FMRpolyG qui colocalisent avec l’ubiquitine et (B) d’un patient NIID où des inclusions intranucléaires de protéine uN2CpolyG colocalisent avec p62.
-
La maladie à inclusions intranucléaires neuronales (NIID) est une maladie neurodégénérative et neuromusculaire qui présente des manifestations cliniques nombreuses et variées : démence, neuropathie périphérique, dystonie neurovégétative, ataxie cérébelleuse, parkinsonisme, tremblement essentiel, convulsions, accident vasculaire cérébral, troubles de la conscience, encéphalopathie, faiblesse des muscles squelettiques, etc. [30]. De même, l’âge d’apparition des premiers symptômes varie grandement et des formes infantiles, juvéniles et adultes ont été décrites. Comme son nom l’indique, cette maladie est caractérisée par la présence d’inclusions intranucléaires éosinophiliques positives à l’ubiquitine et à p62. De façon intéressante, ces inclusions sont strictement identiques à celles décrites dans FXTAS et l’OPDM [31]. Dans la NIID, ces agrégats sont présents dans les neurones et astrocytes des systèmes nerveux central, périphérique et autonome ainsi que dans les cellules de divers autres organes comme la peau ou les muscles squelettiques [32]. La cause génétique de cette maladie a été récemment identifiée comme étant une expansion comprise entre 60 et 200-300 répétitions de tri-nucléotides CGG située dans la région 5’UTR du gène NOTCH2NLC (Figure 2) [6, 30, 33, 34]. Cette expansion de répétitions est également retrouvée chez des patients initialement diagnostiqués pour l’OPDM3, soulignant un continuum entre la NIID et l’OPDM (Figure 4) [8]. Au point de vue moléculaire, il a été montré récemment que les expansions de répétitions CGG situées dans la région 5’UTR du gène NOTCH2NLC sont présentes dans une petite ORF, située en amont de l’ORF principale, et sont donc comme dans FXTAS traduites en une protéine toxique composée de polyglycines, et que nous avons appelée uN2CpolyG [35]. L’expression de cette protéine dans des modèles cellulaires et animaux reproduit la formation d’inclusions intranucléaires typiques de NIID. De plus, des anticorps dirigés contre cette protéine uN2CpolyG confirment sa présence dans les agrégats intranucléaires et ceci, à la fois dans des biopsies de peau et sur des lames de cerveau de patients NIID (Figure 3B). Enfin, des souris exprimant cette protéine présentent des altérations locomotrices, une ataxie et un parkinsonisme associés à une mort neuronale accompagnée de signes de neuroinflammation [35, 36]. Ces résultats suggèrent que la traduction des expansions de répétitions CGG situées dans le gène NOTCH2NLC en une nouvelle protéine composée de polyglycines est la cause de la maladie à inclusions intranucléaires neuronales.

Figure 4 Continuum possible entre les maladies à expansion de répétitions CGG. FXTAS, NIID et les OPDM pourraient former un continuum où les tissus neuronaux et musculaires seraient atteints selon le profil d’expression du gène hôte des répétitions CGG.
Mécanisme d’anticipation dans l’OPDM ?
Dans la DM1, plus les expansions de répétitions CTG sont grandes, plus la maladie est sévère et plus les premiers symptômes de ces pathologies se développent tôt chez les patients. Cette augmentation du nombre de répétitions peut se produire lors de la méiose et, donc, lors du passage d’une génération à la suivante. Ce phénomène conduit ainsi à un mécanisme d’anticipation où les enfants atteints sont généralement porteurs d’un nombre de répétitions plus élevé que leur parent et, par conséquent, développent des manifestations cliniques plus sévères et plus précoces [37]. Toutefois, dans les maladies causées par des expansions de répétitions CGG situées dans des régions 5’UTR, l’existence d’un mécanisme d’anticipation et/ou une corrélation exacte entre la taille de l’expansion et la sévérité des symptômes ne font pas actuellement consensus [7]. En effet, certains individus étaient porteurs de longues expansions de répétitions CGG dans les gènes LRP12, GIPC1 ou NOTCH2NLC mais ne développaient pas les symptômes de l’OPDM [2, 38, 39]. Par exemple, dans l’OPDM2, un individu asymptomatique était porteur d’une expansion de plus de 500 répétitions CGG dans le gène GIPC1 alors que ses deux enfants sont porteurs d’expansions de répétitions CGG plus courtes (117 et 113 répétitions) et ont tous deux développé des formes classiques d’OPDM [2]. Cette longue expansion de 500 répétitions CGG est méthylée, conduisant à une diminution d’expression de la protéine GIPC1 portée par cet allèle, suggérant qu’un mécanisme de perte de fonction est peu probable dans l’OPDM2. De manière similaire, des patients possédant un nombre de répétitions CGG supérieur à 300 ne développent pas de symptômes particuliers contrairement à leurs enfants porteurs d’expansions plus courtes et qui développent une NIID et/ou une OPDM3. Il a été montré récemment que ces longues expansions (200-300 répétitions CGG) conduisent à des modifications épigénétiques du promoteur et donc à une perte d’expression du gène et de la protéine NOTCH2NLC, suggérant ainsi qu’un mécanisme de perte de fonction est peu probable dans la NIID et l’OPDM3 [38].
Ces données sont à mettre en perspective avec la physiopathologie du FXS et du FXTAS. En effet, les expansions de répétitions CGG situées dans le gène FMR1 peuvent conduire à une perte de fonction de la protéine FMRP dans FXS, lorsque l’expansion CGG est longue et méthylée, ou à la génération d’une nouvelle protéine toxique FMRpolyG dans FXTAS, lorsque l’expansion CGG est plus courte, non méthylée et donc transcrite. Bien que présentant une séquence et une localisation identiques, les répétitions CGG sont donc responsables de deux maladies différentes, FXS ou FXTAS, selon deux mécanismes de toxicité distincts qui sont dépendants de la taille de cette expansion. Ces informations ont plusieurs conséquences importantes. Premièrement, ces données montrent que l’haploinsuffisance des protéines LRP12, GIPC1, NOTCH2NLC et RILPL1 due à de longues expansions CGG, ne conduirait pas aux maladies NIID et OPDM. La diminution d’expression de ces protéines peut probablement être compensée par le second allèle, ce qui n’est pas le cas pour la protéine FMRP puisque le gène FMR1 est porté par le chromosome X et l’absence de la protéine FMRP est donc complète chez les hommes, conduisant ainsi au syndrome de l’X fragile [24]. Deuxièmement, des expansions supérieures à 200-300 répétitions CGG pourraient entraîner un effet protecteur puisque ces expansions seraient alors méthylées, conduisant ainsi à une inhibition de la transcription du gène hôte de ces répétitions et, par conséquent, une absence de leur traduction en protéines toxiques composées de polyglycines.
Un phénomène d’anticipation n’a pas été formellement observé chez les patients atteints d’OPDM. De plus, la transmission de cette pathologie n’est pas systématiquement dominante en raison des phénomènes d’expansion et/ou de contraction aléatoire des répétitions CGG. Ceci pourrait expliquer la complexité d’interprétation des arbres généalogiques dans ces familles. En effet, les protéines à polyglycines sont toxiques à partir d’une cinquantaine de glycines mais, au-dessus de 300 répétitions CGG, ces expansions peuvent être méthylées conduisant à une absence de transcription et, donc, de traduction en protéines polyglycines toxiques. Ces données suggèrent une fenêtre pathologique étroite pour ces expansions de répétitions CGG.
Conclusion : quels mécanismes de toxicité pour les expansions de répétitions CGG dans l’OPDM ?
Il est important de noter que ces mutations sont similaires en taille et en séquence (expansions de 60 à 200-300 répétitions CGG) mais localisées dans des gènes différents, suggérant ainsi que les expansions de répétitions CGG jouent un rôle essentiel dans la pathogenèse de l’OPDM et ceci indépendamment de leur gène hôte. Cette hypothèse est renforcée par une absence de méthylation des promoteurs des gènes LRP12, GIPC1, NOTCH2NLC et RILPL1 ainsi que par des niveaux d’expression ARN et protéique de ces gènes inchangés chez les patients atteints d’OPDM, excluant ainsi un mécanisme de perte de fonction dans cette maladie [2, 9, 38, 39]. Il reste donc à déterminer le mécanisme de toxicité de ces répétitions CGG. À ce titre, la similarité en taille, en séquence et en localisation des mutations responsables de l’OPDM, ainsi que la présence d’inclusions intranucléaires positives à p62 et l’ubiquitine identiques chez les patients atteints d’OPDM, et ceci quel que soit le gène contenant ces répétitions CGG, n’est pas sans rappeler deux autres maladies : FXTAS et NIID. Dans ces pathologies, ces expansions de répétitions CGG, bien que localisées dans des régions prédites initialement comme non-codantes, sont traduites en protéines composées de polyglycines qui sont toxiques et qui forment des inclusions intranucléaires typiques de celles observées chez ces patients [37]. On peut se demander si, comme dans NIID et FXTAS, les expansions de répétitions CGG responsables de l’OPDM ne sont pas elles aussi traduites en nouvelles protéines composées de polyglycines, qui formeraient alors les inclusions intranucléaires caractéristiques observées dans les fibres musculaires de ces patients. Cette hypothèse est particulièrement attrayante compte tenu de l’observation récente de la traduction des répétitions CGG en une protéine polyglycine toxique dans NIID [37], et la découverte que cette mutation est également responsable de l’OPDM3 [8], suggérant ainsi fortement un continuum et un mécanisme de toxicité identique entre ces différentes pathologies.
Si un mécanisme commun existait entre ces différentes pathologies neurodégénératives et neuromusculaires, des stratégies thérapeutiques communes dirigées contre les répétitions de nucléotides CGG et/ou les protéines composées de polyglycines pourraient être envisagées. En effet, la découverte récente de molécules pharmacologiques et/ou d’oligonucléotides antisens qui peuvent lier spécifiquement les expansions de répétitions CGG et ainsi diminuer l’expression toxique de FMRpolyG dans des modèles cellulaires et animaux de FXTAS [40], pourrait apporter un espoir thérapeutique pour l’ensemble des maladies à polyglycines.
Liens d’intérêt
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les données publiées dans cet article.
Remerciements
Ce travail a été soutenu par les financements suivants : FDT202001010855 (MB); FRM EQU202103012936 (NCB); ANR-10-LABX0030-INRT and ANR-10-IDEX-0002-02 (IGBMC).
Références
- Satoyoshi E, Kinoshita M. Oculopharyngodistal myopathy. Arch Neurol 1977 Feb ; 34 (2) : 89–92. [CrossRef] [PubMed] [Google Scholar]
- Deng J, Yu J, Li P, et al. Expansion of GGC Repeat in GIPC1 Is Associated with Oculopharyngodistal Myopathy. Am J Hum Genet 2020 Jun 4 ; 106 (6) : 793–804. [CrossRef] [PubMed] [Google Scholar]
- Durmus H, Laval SH, Deymeer F, et al. Oculopharyngodistal myopathy is a distinct entity: clinical and genetic features of 47 patients. Neurology 2011 Jan 18 ; 76 (3) : 227–35. [CrossRef] [PubMed] [Google Scholar]
- Zhao J, Liu J, Xiao J, et al. Clinical and muscle imaging findings in 14 mainland chinese patients with oculopharyngodistal myopathy. PLoS One 2015 Jun 3 ; 10 (6) : e0128629. [CrossRef] [PubMed] [Google Scholar]
- Ogasawara M, Iida A, Kumutpongpanich T, et al. CGG expansion in NOTCH2NLC is associated with oculopharyngodistal myopathy with neurological manifestations. Acta Neuropathol Commun 2020 Nov 25 ; 8 (1) : 204. [CrossRef] [PubMed] [Google Scholar]
- Ishiura H, Shibata S, Yoshimura J, et al. Noncoding CGG repeat expansions in neuronal intranuclear inclusion disease, oculopharyngodistal myopathy and an overlapping disease. Nat Genet 2019 Aug ; 51 (8) : 1222–232. [CrossRef] [PubMed] [Google Scholar]
- Xi J, Wang X, Yue D, et al. 5’ UTR CGG repeat expansion in GIPC1 is associated with oculopharyngodistal myopathy. Brain 2021 Mar 3 ; 144 (2) : 601–14. [CrossRef] [PubMed] [Google Scholar]
- Yu J, Deng J, Guo X, et al. The GGC repeat expansion in NOTCH2NLC is associated with oculopharyngodistal myopathy type 3. Brain 2021 Jul 28 ; 144 (6) : 1819–32. [CrossRef] [PubMed] [Google Scholar]
- Yu J, Shan J, Yu M, et al. The CGG repeat expansion in RILPL1 is associated with oculopharyngodistal myopathy type 4. Am J Hum Genet 2022 Mar 3 ; 109 (3) : 533–41. [CrossRef] [PubMed] [Google Scholar]
- Lee JE, Cooper TA. Pathogenic mechanisms of myotonic dystrophy. Biochem Soc Trans 2009 Dec ; 37 : 1281–6. [CrossRef] [PubMed] [Google Scholar]
- Liquori CL, Ricker K, Moseley ML, et al. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science 2001 Aug 3 ; 293 (5531) : 864–7. [CrossRef] [PubMed] [Google Scholar]
- Miller JW, Urbinati CR, Teng-Umnuay P, et al. Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. EMBO J 2000 Sep 1 ; 19 (17) : 4439–48. [CrossRef] [PubMed] [Google Scholar]
- Ho TH, Charlet-B N, Poulos MG, et al. Muscleblind proteins regulate alternative splicing. EMBO J 2004 Aug 4 ; 23 (15) : 3103–12. [CrossRef] [PubMed] [Google Scholar]
- Dansithong W, Paul S, Comai L, et al. MBNL1 is the primary determinant of focus formation and aberrant insulin receptor splicing in DM1. J Biol Chem 2005 ; 280 : 5773–80. [CrossRef] [PubMed] [Google Scholar]
- Tomé FM, Fardeau M. Nuclear inclusions in oculopharyngeal dystrophy. Acta Neuropathol 1980 ; 49(1) : 85–7. [CrossRef] [PubMed] [Google Scholar]
- Brais B, Bouchard JP, Xie YG, et al. Short GCG expansions in the PABP2 gene cause oculopharyngeal muscular dystrophy. Nat Genet 1998 Feb ; 18 (2) : 164–7. [CrossRef] [PubMed] [Google Scholar]
- Calado A, Tomé FM, Brais B, et al. Nuclear inclusions in oculopharyngeal muscular dystrophy consist of poly(A) binding protein 2 aggregates which sequester poly(A) RNA. Hum Mol Genet 2000 Sep 22 ; 9 (15) : 2321–8. [CrossRef] [PubMed] [Google Scholar]
- Trollet C, Anvar SY, Venema A, et al. Molecular and phenotypic characterization of a mouse model of oculopharyngeal muscular dystrophy reveals severe muscular atrophy restricted to fast glycolytic fibres. Hum Mol Genet 2010 Jun 1 ; 19 (11) : 2191–207. [CrossRef] [PubMed] [Google Scholar]
- Jacquemont S, Hagerman RJ, Leehey M, et al. Fragile X premutation tremor/ataxia syndrome: molecular, clinical, and neuroimaging correlates. Am J Hum Genet 2003 Apr ; 72 (4) : 869–78. [CrossRef] [PubMed] [Google Scholar]
- Greco CM, Hagerman RJ, Tassone F, et al. Neuronal intranuclear inclusions in a new cerebellar tremor/ataxia syndrome among fragile X carriers. Brain 2002 Aug ; 125 : 1760–71. [CrossRef] [PubMed] [Google Scholar]
- Greco CM, Berman RF, Martin RM, et al. Neuropathology of fragile X-associated tremor/ataxia syndrome (FXTAS). Brain 2006 Jan ; 129 : 243–55. [CrossRef] [PubMed] [Google Scholar]
- Hunsaker MR, Greco CM, Spath MA, et al. Widespread noncentral nervous system organ pathology in fragile X premutation carriers with fragile X-associated tremor/ataxia syndrome and CGG knock-in mice. Acta Neuropathol 2011 Oct ; 122 (4) : 467–79. [CrossRef] [PubMed] [Google Scholar]
- Hagerman RJ, Leehey M, Heinrichs W, et al. Intention tremor, parkinsonism, and generalized brain atrophy in male carriers of fragile X. Neurology 2001 Jul 10 ; 57 (1) : 127–30. [CrossRef] [PubMed] [Google Scholar]
- Oberlé I, Rousseau F, Heitz D, et al. Instability of a 550-base pair DNA segment and abnormal methylation in fragile X syndrome. Science 1991 May 24 ; 252 (5009) : 1097–102. [CrossRef] [PubMed] [Google Scholar]
- Coffee B, Zhang F, Warren ST, et al. Acetylated histones are associated with FMR1 in normal but not fragile X-syndrome cells. Nat Genet 1999 May ; 22 (1) : 98–101. [CrossRef] [PubMed] [Google Scholar]
- Tassone F, Hagerman RJ, Loesch DZ, et al. Fragile X males with unmethylated, full mutation trinucleotide repeat expansions have elevated levels of FMR1 messenger RNA. Am J Med Genet 2000 Sep 18 ; 94 (3) : 232–6. [CrossRef] [PubMed] [Google Scholar]
- Todd PK, Oh SY, Krans A, et al. CGG repeat-associated translation mediates neurodegeneration in fragile X tremor ataxia syndrome. Neuron 2013 May 8 ; 78 (3) : 440–55. [CrossRef] [PubMed] [Google Scholar]
- Sellier C, Buijsen RAM, He F, et al. Translation of Expanded CGG Repeats into FMRpolyG Is Pathogenic and May Contribute to Fragile X Tremor Ataxia Syndrome. Neuron 2017 Jan 18 ; 93 (2) : 331–47. [CrossRef] [PubMed] [Google Scholar]
- Kozak M. Context effects and inefficient initiation at non-AUG codons in eucaryotic cell-free translation systems. Mol Cell Biol 1989 Nov ; 9 (11) : 5073–80. [PubMed] [Google Scholar]
- Sone J, Mitsuhashi S, Fujita A, et al. Long-read sequencing identifies GGC repeat expansions in NOTCH2NLC associated with neuronal intranuclear inclusion disease. Nat Genet 2019 Aug ; 51 (8) : 1215–21. [CrossRef] [PubMed] [Google Scholar]
- Gelpi E, Botta-Orfila T, Bodi L, et al. Neuronal intranuclear (hyaline) inclusion disease and fragile X-associated tremor/ataxia syndrome: a morphological and molecular dilemma. Brain 2017 Aug 1 ; 140 (8) : e51. [CrossRef] [PubMed] [Google Scholar]
- Kimber TE, Blumbergs PC, Rice JP, et al. Familial neuronal intranuclear inclusion disease with ubiquitin positive inclusions. J Neurol Sci 1998 Sep 18 ; 160 (1) : 33–40. [CrossRef] [PubMed] [Google Scholar]
- Deng J, Gu M, Miao Y, et al. Long-read sequencing identified repeat expansions in the 5’UTR of the NOTCH2NLC gene from Chinese patients with neuronal intranuclear inclusion disease. J Med Genet 2019 Nov ; 56 (11) : 758–64. [CrossRef] [PubMed] [Google Scholar]
- Tian Y, Wang JL, Huang W, et al. Expansion of HumanSpecific GGC Repeat in Neuronal Intranuclear Inclusion DiseaseRelated Disorders. Am J Hum Genet 2019 Jul 3 ; 105 (1) : 166–76. [CrossRef] [PubMed] [Google Scholar]
- Boivin M, Deng J, Pfister V, et al. Translation of GGC repeat expansions into a toxic polyglycine protein in NIID defines a novel class of human genetic disorders: The polyG diseases. Neuron 2021 Jun 2 ; 109 (11) : 1825–35. [CrossRef] [PubMed] [Google Scholar]
- Zhong S, Lian Y, Luo W, et al. Upstream open reading frame with NOTCH2NLC GGC expansion generates polyglycine aggregates and disrupts nucleocytoplasmic transport: implications for polyglycine diseases. Acta Neuropathol 2021 Oct 25. [PubMed] [Google Scholar]
- Boivin M, Charlet-Berguerand N. Trinucleotide CGG Repeat Diseases: An Expanding Field of Polyglycine Proteins ? Front Genet 2022 Feb 28 ; 13 : 843014. [CrossRef] [PubMed] [Google Scholar]
- Fukuda H, Yamaguchi D, Nyquist K, et al. Father-to-offspring transmission of extremely long NOTCH2NLC repeat expansions with contractions: genetic and epigenetic profiling with long-read sequencing. Clin Epigenetics 2021 Nov 13 ; 13 (1) : 204. [CrossRef] [PubMed] [Google Scholar]
- Kumutpongpanich T, Ogasawara M, Ozaki A, et al. Clinicopathologic Features of Oculopharyngodistal Myopathy With LRP12 CGG Repeat Expansions Compared With Other Oculopharyngodistal Myopathy Subtypes. JAMA Neurol 2021 Jul 1 ; 78 (7) : 853–63. [CrossRef] [PubMed] [Google Scholar]
- Xu K, Li Y, Allen EG, et al. Therapeutic Development for CGG Repeat Expansion-Associated Neurodegeneration. Front Cell Neurosci 2021 May ; 12 (15) : 655568. [CrossRef] [PubMed] [Google Scholar]
Liste des figures
|
Figure 1 Caractéristiques histopathologiques de biopsies de patients atteints d’OPDM. (A, B) Présence de vacuoles cytoplasmiques révélées par une coloration (A) au trichrome de Gomori ou (B) à l’hématoxyline-éosine et (C) d’inclusions intranucléaires positives pour la protéine p62. |
| Dans le texte | |
|
Figure 2 Les expansions de répétitions CGG impliquées dans des pathologies. Des expansions de répétitions CGG identifiées dans les régions 5’UTR des gènes FMR1, LRP12, GIPC1, NOTCH2NLC et RILPL1 sont responsables du syndrome de l’X fragile (FXS) ou du syndrome de tremblement et d’ataxie lié à l’X fragile (FXTAS), de la maladie à inclusions intranucléaires neuronales (NIID) et de la myopathie oculo-pharyngo-distale (OPDM). |
| Dans le texte | |
|
Figure 3 Inclusions intranucléaires dans FXTAS et NIID. Immunofluorescence sur des lames de cervelet (A) d’un patient FXTAS montrant des inclusions intranucléaires de protéine FMRpolyG qui colocalisent avec l’ubiquitine et (B) d’un patient NIID où des inclusions intranucléaires de protéine uN2CpolyG colocalisent avec p62. |
| Dans le texte | |
|
Figure 4 Continuum possible entre les maladies à expansion de répétitions CGG. FXTAS, NIID et les OPDM pourraient former un continuum où les tissus neuronaux et musculaires seraient atteints selon le profil d’expression du gène hôte des répétitions CGG. |
| Dans le texte | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.